Arq. Bras. Oftalmol. 2000;63 (6 )
:463-468
| DOI: 10.1590/S0004-27492000000600007
Abstract
Introdução: Para se verificar a prevalência de anomalias oculares em indivíduos portadores de deficiência auditiva de causa genética definitiva ou suspeita, este trabalho apresenta a avaliação oftalmológica de 97 indivíduos portadores de deficiência auditiva. Pacientes e Métodos: 97 indivíduos com diagnóstico definitivo ou suspeito de causa genética para disacusia foram submetidos a exame clínico oftalmológico completo; destes, 10 foram excluídos. Resultados: 42 (48,28%) dos pacientes apresentaram uma ou mais anomalias oculares, 22 (25,29%) pacientes apresentaram várias anormalidades oculares e quadro clínico compatíveis com síndromes genéticas estabelecidas. Conclusões: O exame oftalmológico é importante no diagnóstico sindrômico e etiológico de alguns quadros de disacusia, pois as alterações oculares podem ser a única anomalia associada à mesma.
Keywords: Anomalias oculares; Deficiência auditiva genética
Arq. Bras. Oftalmol. 2002;65 (4 )
:419-426
| DOI: 10.1590/S0004-27492002000400005
Abstract
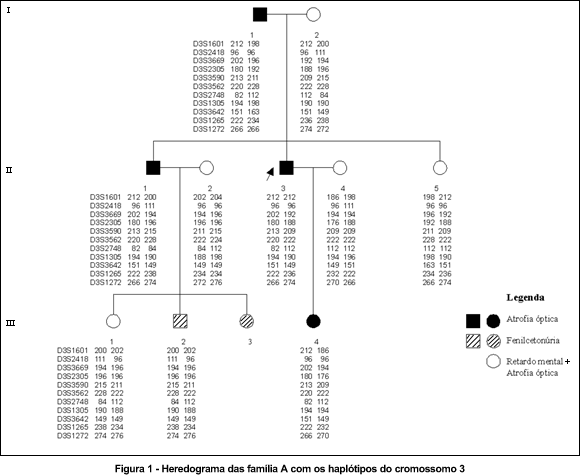
Objetivos: A atrofia óptica autossômica dominante, tipo Kjer ou juvenil, é neuropatia óptica hereditária que causa perda de acuidade visual, anormalidades da visão de cores e defeitos do campo visual, caracterizada por palidez do disco óptico. O gene desta doença foi mapeado por análise de ligação genética em um intervalo de 1,4 cM no cromossomo 3q28-29 entre os marcadores microssatélites D3S3669 e D3S3562. Embora a maioria das famílias estudadas tenha mostrado ligação para a região cromossômica 3q28-29, uma família foi mapeada no cromossomo 18q12.2-12.3. Este trabalho analisa a ligação da atrofia óptica em três famílias com marcadores polimórficos para os cromossomos 3q28-29 e 18q12.2-12.3. Métodos: Cinqüenta e sete indivíduos de três famílias foram submetidos a exame oftalmológico e coleta de sangue. O DNA foi extraído e amplificado em reações de polimerase em cadeia (PCR) com marcadores polimórficos para os cromossomos 3q28-29 e 18q12.2-12.3. Os fragmentos de PCR foram mensurados em seqüenciador automático (373 DNA sequencer). Estes números foram utilizados como alelos para análise de haplótipos. Os "lod scores" foram calculados pelo programa MLINK. Resultados: Na primeira família houve suspeita da atrofia óptica mapear para o cromossomo 3q28-29, mas sem significância estatística no valor do "lod score". Na segunda família a atrofia óptica apresentou ligação para este locus. Os eventos de recombinação nesta família localizaram o gene num intervalo de 2 cM entre os marcadores D3S3669 e D3S2305. O "lod score" máximo obtido foi de 3,56 no theta de 0,00 com o marcador D3S3669. A terceira família não apresentou ligação nos cromossomos 3q28-29 e 18q12.2-12.3. Conclusão: O fato da terceira família não mapear para nenhum dos dois loci já descritos é indicativo de que existe heterogeneidade genética na atrofia óptica autossômica dominante e levanta a possibilidade de existir um terceiro locus para esta doença.
Keywords: Atrofia óptica autossômica dominante; Acuidade visual; Campos visuais; Defeitos da visão de cores; Ligação (Genética)
Arq. Bras. Oftalmol. 2002;65 (6 )
:623-628
| DOI: 10.1590/S0004-27492002000600005
Abstract
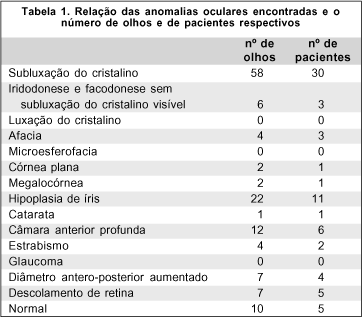
Objetivo: Para se identificar as alterações oculares presentes na síndrome de Marfan, este trabalho apresenta a avaliação oftalmológica de 46 indivíduos portadores desta doença. Métodos: Estudo prospectivo com avaliação clínica de 46 pacientes portadores de síndrome de Marfan com avaliação oftalmológica completa. Dezessete pacientes também foram submetidos a exame genético clínico e estudo molecular. Resultados: Dos quarenta e seis pacientes incluídos neste estudo, as seguintes alterações oculares foram encontradas com maior freqüência: subluxação do cristalino (67,3%), hipoplasia de íris (23,9%), descolamento de retina (7,6%), córnea plana (2,2%), megalocórnea (2,2%) e miopia ou astigmatismo miópico (34,8%). Cinco pacientes (10,9%) apresentaram exame ocular normal em ambos os olhos. Detectou-se uma mutação patogênica distinta das relatadas na literatura em uma paciente, uma mutação de sentido trocado que ocorreu no éxon 28 levando à mudança de aminoácido C1166Y. Conclusões: As alterações oculares da síndrome de Marfan são freqüentes e o conhecimento do gene responsável FBN-1 e de sua expressão no olho auxiliam para o diagnóstico e tratamento destas anomalias.
Keywords: Síndrome de Marfan; Anomalias do olho; Oftalmopatias
Arq. Bras. Oftalmol. 2003;66 (5 )
:579-581
| DOI: 10.1590/S0004-27492003000500007
Abstract
OBJETIVO: Analisar a incidência, evolução clínica, alteração oftalmológica e prognóstico de vida de pacientes com hemorragia subaracnóidea e síndrome de Terson. MÉTODOS: Estudo prospectivo e consecutivo de pacientes admitidos no pronto socorro de neurocirurgia da Universidade Federal de São Paulo com diagnóstico de hemorragia subaracnóidea aguda. Após exame neurológico, o mapeamento de retina foi realizado em todos os pacientes na admissão e no 3º, 7º, 30º e 60º dia. Em todos os casos foi realizada a correlação entre a escala de Hunt e Hess e a presença de hemorragia intra-ocular. RESULTADOS: Dezessete pacientes foram examinados durante julho a outubro de 2000. A síndrome de Terson foi observada em 5 casos (29,4%). Em 15 pacientes a etiologia da hemorragia foi ruptura de aneurisma cerebral e em 2 casos a causa foi relacionada a traumatismo crânio-encefálico. Não houve predominância significante de sexo (9F e 8M) e a idade mediana foi de 48 anos (22 a 80 anos). Houve 4 óbitos de pacientes com síndrome de Terson e apenas 1 no grupo de pacientes sem alteração ocular. Não houve nenhuma correlação entre a gravidade do quadro clínico e a presença da síndrome de Terson. CONCLUSÃO: Neste estudo, a incidência da síndrome de Terson foi de 29,4% e sua presença indicou alto risco de mortalidade (80% dos casos com a síndrome de Terson).
Keywords: Aneurisma cerebral; Trauma craniocerebral; Síndrome; Corpo vítreo; Hemorragia vítrea; Hemorragia subaracnóidea; Hemorragia retiniana; Estudos prospectivos
Arq. Bras. Oftalmol. 2006;69 (4 )
:589-592
| DOI: 10.1590/S0004-27492006000400025
Abstract
O termo fundus flavimaculatus (doença de Stargardt) descreve um grupo de distrofias maculares hereditárias caracterizadas por múltiplos "flecks" amarelados em nível do epitélio pigmentar da retina. Os autores descrevem os achados de tomografia de coerência óptica (OCT) em paciente portador de doença de Stargardt e sugerem que a OCT tem validade como exame subsidiário no estudo das características da retina de pacientes portadores da doença de Stargardt, embora estudos envolvendo maior número de pacientes sejam indicados para permitir traçar-se o perfil das alterações mais comuns nestes casos.
Keywords: Degeneração macular; Tomografia de coerência óptica; Pigmentos da retina; Fundus oculi; Relatos de casos [tipo de publicação]
Arq. Bras. Oftalmol. 2007;70 (5 )
:739-745
| DOI: 10.1590/S0004-27492007000500003
Abstract
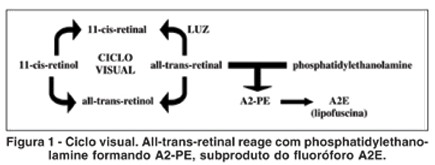
OBJETIVOS: Definir características do exame de autofluorescência, verificando sua utilidade no diagnóstico e acompanhamento de distrofias retinianas. MÉTODOS: Participaram do estudo, 28 pacientes, adultos, divididos igualmente em quatro grupos com diagnósticos de doença de Stargardt, distrofia de Cones, retinose pigmentar e voluntários saudáveis para estabelecimento do padrão de normalidade. Em média foram obtidas nove imagens com o filtro para angiofluoresceinografia para a formação da imagem autofluorescente no Heidelberg Retina Angiograph2. As imagens de cada grupo de pacientes foram analisadas para verificar características comuns. RESULTADOS: As imagens fundoscópicas autofluorescentes dos voluntários do grupo controle mostraram área foveal hipoautofluorescente em relação à retina do pólo posterior. As imagens dos portadores de doença de Stargardt, em geral, apresentaram lesão hipoautofluorescente, correspondendo à área macular. As principais alterações da autofluorescência em pacientes com distrofia de cones foram hipoautofluorescência macular com halo hiperautofluorescente. Nos portadores de retinose pigmentar, foram encontrados pigmentos periféricos causando hipoautofluorescência. Na região macular, hipoautofluorescência ou apenas desorganização do pigmento. CONCLUSÃO: O estudo mostrou a existência de padrões de autofluorescência de fundo nas distrofias de retina que permitem o diagnóstico e melhor interpretação da fisiopatogenia destas doenças.
Keywords: Doenças retinianas; Lipofuscina; Epitélio pigmentado ocular; Oftalmopatias hereditárias; Angiofluoresceinografia
Arq. Bras. Oftalmol. 2008;71 (6 )
:799-804
| DOI: 10.1590/S0004-27492008000600006
Abstract
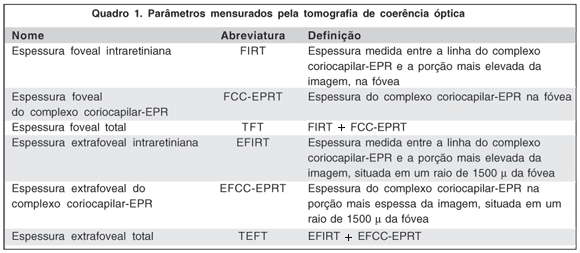
OBJETIVO: Identificar os achados na tomografia de coerência óptica (OCT) e suas variações ao longo de 12 meses, em pacientes portadores de degeneração macular relacionada à idade (DRMI), submetidos à terapia fotodinâmica com verteporfina (TFD). DESENHO DO ESTUDO: Série de casos, aberto, não aleatório e intervencionista. MÉTODOS: Pacientes acima de 50 anos, portadores de DMRI neovascular foram submetidos ao exame oftalmológico completo, angiofluoresceinografia e OCT antes do início do tratamento (V0) e 3, 6, 9 e 12 meses após (V3, V6, V9 e V12, respectivamente). O tratamento empregado foi a TFD. A acuidade visual (AV) foi mensurada usando-se a tabela do ETDRS. Realizaram-se as medições das espessuras foveais: espessura intraretiniana foveal (FIRT), espessura foveal do complexo coriocapilar - EPR (FCC-EPRT) e espessura foveal total (TFT). Realizaram-se as mensurações das espessuras extrafoveais, em um raio de 1500 µ da fóvea: espessura intraretiniana extrafoveal (EFIRT), espessura extrafoveal do complexo coriocapilar - EPR (EFCC-EPRT) e espessura extrafoveal total (TEFT). Análise estatística foi realizada usando-se a análise de variância em blocos. RESULTADOS: Vinte e três olhos de 23 pacientes foram avaliados. Foram identificados nove achados à OCT: 1º) espessamento das camadas intraretinianas na fóvea; 2º) espessamento das camadas intraretinianas na região extrafoveal; 3º) espessamento do complexo coriocapilar-EPR (FCC-EPRT) na fóvea; 4º) espessamento do complexo coriocapilar-EPR na área extrafoveal; 5º) presença de fluido sub-EPR; 6º) presença de fluido sub-retiniano; 7º) presença de fluido intraretiniano; 8º) presença da membrana hialóide posterior aderida à retina; 9º) presença da depressão foveal. Na visita inicial a FIRT e a TFT foram respectivamente 398,5 µ e 639,2 µ. Em V12 foram 173,7 µ e 423,9 µ. A variação foi estatisticamente significante (p = 0,008 e p = 0,003, respectivamente). As outras espessuras mensuradas não tiveram variação estatisticamente significante. A depressão foveal estava presente em 36,4% dos olhos na visita inicial e em 78,3% em V12. O fluido sub-retiniano estava presente em 36,4% em V0 e em 8,7% em V12. A AV na visita inicial foi 0,93 e na visita final foi 1,04 (p = 0,127). CONCLUSÕES: A AV permaneceu inalterada ao longo do estudo. A depressão foveal estava presente em 78,3% dos olhos em V12. A diminuição da FIRT e da TFT foi estatisticamente significante entre V0 e V12.
Keywords: Degeneração macular; Efeito idade; Tomografia de coerência óptica; Fotoquimioterapia; Agentes fotossensibilizantes
Arq. Bras. Oftalmol. 2009;72 (4 )
:560-566
| DOI: 10.1590/S0004-27492009000400026
Abstract
As distrofias hereditárias de retina abrangem um amplo número de doenças caracterizadas por lenta e progressiva degeneração da retina. São o resultado de mutações em genes expressos em fotorreceptores e no epitélio pigmentado da retina. A herança pode ser autossômica dominante, autossômica recessiva, ligada ao X recessiva, digênica ou herança mitocondrial. Atualmente não há tratamento para essas doenças e os pacientes convivem com a perda progressiva da visão. O aconselhamento genético e o suporte para reabilitação têm indicação nestes casos. Pesquisas envolvendo a base molecular e genética dessas doenças está continuamente em expansão e ampliam as perspectivas para novas formas de tratamento. Dessa forma, a terapia gênica, que consiste na inserção de material genético exógeno em células de um indivíduo com finalidade terapêutica, tem sido a principal forma de tratamento para as distrofias hereditárias de retina. O olho é um órgão peculiar para a terapia gênica, pois é anatomicamente dividido em compartimentos, imunologicamente privilegiado e com meios transparentes. A maioria das doenças oculares tem defeitos em genes conhecidos. Além disso, há modelo animal bem caracterizado para algumas condições. Propostas para pesquisa clínica em terapia gênica nas degenerações retinianas hereditárias com defeito no gene RPE65, recentemente tiveram aprovação ética e os resultados preliminares obtidos trouxeram grandes expectativas na melhora da qualidade de vida dos pacientes.
Keywords: Retina; Oftalmopatias hereditárias; Terapia de genes; Vetores genéticos; Técnicas de transferência de genes
Arq. Bras. Oftalmol. 2011;74 (5 )
:338-342
| DOI: 10.1590/S0004-27492011000500006
Abstract
Objetivo: Comparar o efeito anti-angiogênico in vitro do Bevacizumab e do Rani bizumab. Métodos: Células endotelias venosas de cordão umbilical (ECV304), cultivadas em meio F12 com adição de 10% de soro fetal bovino, foram plaqueadas e tratadas com concentrações clinicamente relevantes de Bevacizumab e Ranibizumab. As drogas foram administradas logo após risco realizado no meio da cultura (metodologia de scratch). Medidas lineares do espaço livre de proliferação celular foram realizadas 24, 48 e 72 horas após o momento da realização do risco. Todos os experimentos foram realizados em triplicata e a análise estatística foi feita pelo teste T-student. Resultados: O efeito inibitório foi observado em ambas as drogas, apenas nas concentrações 0,5 e 0,7 mg/ml. Na concentração 0,7 mg/ml, o Ranibizumab demonstrou efeito inibitório maior do que o Bevacizumab. Na mesma concentração, o Ranibizumab foi três vezes mais potente que o Bevacizumab. O efeito inibitório foi observado apenas nas primeiras 24 horas para ambas as drogas. Conclusão: O Ranibizumab demonstrou efeito maior quando comparado com o Bevacizumab, porém tal efeito está mais relacionado à diferença na razão molar das drogas do que relacionada com uma diferença real no efeito anti-proliferativo.
Keywords: Neovascularização de coróide; Degeneração macular; Inibidores da angiogênese; Anticorpos monoclonais; Tomografia de coerência óptica; Angiofluoresceinografia; Injeções intravítreas; Corpo vítreo
Arq. Bras. Oftalmol. 2012;75 (3 )
:210-212
| DOI: 10.1590/S0004-27492012000300013
Abstract
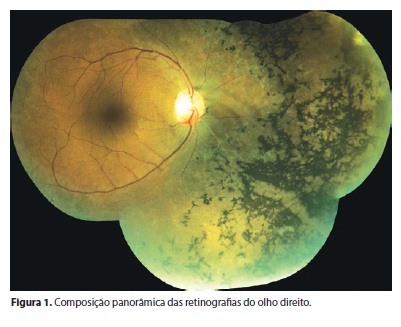
A retinose pigmentada constitui um grupo de doenças causadas por alterações genéticas que levam à degeneração progressiva dos fotorreceptores, principalmente bastonetes. Em geral, tem apresentação bilateral. Este estudo é um relato de caso de uma paciente com acometimento unilateral da retina, de características semelhantes às da retinose pigmentada, com história de trauma ocular antigo. Descrevem-se sua história clínica e achados oftalmológicos.
Keywords: Retinite pigmentosa; Traumatismos oculares; Distrofias retinianas; Relatos de casos
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.


05-tab01tb.jpg)
03-fig01.jpg)
15-fig01.jpg)