Arq. Bras. Oftalmol. 2002; 65 (4): 10.1590/S0004-27492002000400005
Total: 2708
Juliana Maria Ferraz Sallum1; Michel Eid Farah1; Irene Hussels Maumenee2
DOI: 10.1590/S0004-27492002000400005
RESUMO
Objetivos: A atrofia óptica autossômica dominante, tipo Kjer ou juvenil, é neuropatia óptica hereditária que causa perda de acuidade visual, anormalidades da visão de cores e defeitos do campo visual, caracterizada por palidez do disco óptico. O gene desta doença foi mapeado por análise de ligação genética em um intervalo de 1,4 cM no cromossomo 3q28-29 entre os marcadores microssatélites D3S3669 e D3S3562. Embora a maioria das famílias estudadas tenha mostrado ligação para a região cromossômica 3q28-29, uma família foi mapeada no cromossomo 18q12.2-12.3. Este trabalho analisa a ligação da atrofia óptica em três famílias com marcadores polimórficos para os cromossomos 3q28-29 e 18q12.2-12.3. Métodos: Cinqüenta e sete indivíduos de três famílias foram submetidos a exame oftalmológico e coleta de sangue. O DNA foi extraído e amplificado em reações de polimerase em cadeia (PCR) com marcadores polimórficos para os cromossomos 3q28-29 e 18q12.2-12.3. Os fragmentos de PCR foram mensurados em seqüenciador automático (373 DNA sequencer). Estes números foram utilizados como alelos para análise de haplótipos. Os "lod scores" foram calculados pelo programa MLINK. Resultados: Na primeira família houve suspeita da atrofia óptica mapear para o cromossomo 3q28-29, mas sem significância estatística no valor do "lod score". Na segunda família a atrofia óptica apresentou ligação para este locus. Os eventos de recombinação nesta família localizaram o gene num intervalo de 2 cM entre os marcadores D3S3669 e D3S2305. O "lod score" máximo obtido foi de 3,56 no theta de 0,00 com o marcador D3S3669. A terceira família não apresentou ligação nos cromossomos 3q28-29 e 18q12.2-12.3. Conclusão: O fato da terceira família não mapear para nenhum dos dois loci já descritos é indicativo de que existe heterogeneidade genética na atrofia óptica autossômica dominante e levanta a possibilidade de existir um terceiro locus para esta doença.
Descritores: Atrofia óptica autossômica dominante; Acuidade visual; Campos visuais; Defeitos da visão de cores; Ligação (Genética)
ABSTRACT
Purpose: Autosomal dominant optic atrophy is a hereditary optic neuropathy characterized by progressive visual loss in childhood, color vision anomalies, visual field defects and temporal pallor of the optic disc. This disease has been mapped to a 1.4 cM interval in chromosome 3q28-29 between markers D3S3669 and D3S3562. One family was mapped to chromosome 18q12.2-12.3. Linkage analysis in three families with autosomal dominant optic atrophy with polymorphic DNA markers for chromosome 3q28-29 and 18q12.2-12.3. Methods: 57 individuals from three families underwent ophthalmological examination. Genomic DNA was extracted from blood samples. Linkage analysis was performed between the disease and 11 polymorphic markers around 3q28-qter and 18q12.2-12.3. Polymerase chain reaction (PCR) fragments sizes were identified in a scanner gel using a 373 DNA sequencer. These numbers were used as alleles for pedigree analysis. The lod scores were calculated using the MLINK program. Results: All three families presented optic atrophy with autosomal dominant pattern of inheritance, variable expression and high penetrance. Two families were linked to 3q28-29 markers. A maximal lod score of 3.56, at a recombination fraction of zero, was obtained using the marker D3S3669 in one family. The linkage area was defined in a 2 cM interval by haplotype analysis between markers D3S2418 and D3S1305, because patients III.4 and III.14 showed crossing-overs. The third family was not linked to 3q28-29 neither to 18q12.2-12.3. Conclusions: There is genetic heterogeneity in autosomal dominant optic atrophy, because the third family did not map to any known locus. And a third locus for this disease may exist.
Keywords: Autosomal dominant optic atrophy; Visual acuity; Visual fields; Color vision defects; Linkage (Genetics)
INTRODUÇÃO
Atrofia óptica autossômica dominante é a forma mais comum de atrofia óptica familial; é também denominada juvenil, tipo "Kjer" ou OPA1. Kjer em 1959 definiu esta doença como uma anomalia hereditária caracterizada pelo aparecimento de atrofia óptica bilateral na infância(1). Já foram descritas associações com retardo mental e perdas auditivas neurossensoriais(2-3).
A hereditariedade segue o padrão monogênico autossômico dominante com alta penetrância e expressividade variável (OMIM #165500)(4).
Smith em 1972, fez uma revisão das características clínicas e listou os critérios para o diagnóstico da atrofia óptica dominante(5). No contexto de uma família, na qual a atrofia óptica dominante segrega, os critérios mínimos de diagnóstico são: palidez de disco óptico total ou temporal e qualquer defeito na visão de cores. A acuidade visual pode estar diminuída ou apresentar-se normal. E em apenas metade dos indivíduos afetados o aparecimento ocorre antes dos dez anos(6).
Esta doença afeta primariamente a camada de células ganglionares da retina, com conseqüente atrofia óptica ascendente. Existe atrofia difusa da camada de células ganglionares da retina com aparência normal das camadas nucleares interna e externa; desmielinização não inflamatória no nervo óptico e perda de tecido nervoso do lado temporal do disco(7). Tanto a eletrofisiologia quanto o exame histopatológico sugerem um defeito na camada de células ganglionares. Isto sugere que o gene responsável pela atrofia óptica se expresse nas células ganglionares(8).
O primeiro estudo de ligação em atrofia óptica dominante foi publicado em 1983, numa grande família com atrofia óptica autossômica dominante com evidência de ligação com o antígeno do grupo sanguíneo Kidd(9). Posteriormente a localização genética deste antígeno foi identificada no cromossomo 18q12(10).
Eiberg e cols em 1994 mapearam a atrofia óptica autossômica dominante no cromossomo 3q28-qter em três famílias dinamarquesas. Neste estudo de ligação os autores demonstraram ligação do gene da doença, que eles simbolizaram como OPA1, com o marcador D3S1314 ("lod score" máximo 10,34 em theta = 0,075). A região onde o gene OPA1 mapeava era delimitada pelos marcadores D3S1314 e D3S1265 e media mais de 10 cM(11).
O intervalo de ligação foi refinado para um intervalo de 1,4 cM no cromossomo 3q28-q29, delimitado pelos marcadores D3S3669 e D3S3562 em um segmento de DNA genômico, "YAC contig" de 3.000 kb(12). O gene OPA1 foi localizado no cromossomo 3q28 a 400 kb do marcador D3S1523(13).
Inicialmente acreditava-se não existir evidência de heterogeneidade genética(14). Seller e cols descreveram nove famílias, das quais uma não apresentou ligação para este locus, o que levantou a possibilidade de heterogeneidade genética(15). A heterogeneidade genética só foi comprovada quando Kerrison e cols(16) demonstraram ligação da atrofia óptica autossômica dominante, com marcadores do cromossomo 18q12.2-12.3, na família que anteriormente havia apresentado ligação com o grupo sangüíneo Kidd(9). A análise de múltiplos pontos demonstrou "lod score" maior que 3, numa região de aproximadamente 3 cM, delimitada por D18S34 e D18S479.
Vale ressaltar que mais de 56 heredogramas com atrofia óptica já foram descritos com ligação no locus 3q28-29 e apenas um com ligação no 18q12.2-12.3, portanto parece que a maioria dos casos está relacionada ao gene do cromossomo 3.
O gene homólogo no homem do gene Drosophila "Hairy" (HRY; OMIM*139605; Genbank: L19314) e o gene da diacilglicerol quinase gama (DGKG; OMIM* 601854; Genbank: D26135) já foram pesquisados e excluídos como genes candidatos a responsáveis pela atrofia óptica autossômica dominante(5,17-19). Até o presente momento ainda não se conseguiu identificar o gene responsável pela atrofia óptica autossômica dominante (ver nota do autor).
Este trabalho objetivou fazer um estudo genético clínico da atrofia óptica autossômica dominante em três famílias com esta doença; observar se o gene responsável por esta doença nestas famílias está ligado aos marcadores polimórficos da região 3q28-29; e caso não se detecte ligação neste locus, verificar se existe heterogeneidade genética testando também marcadores no cromossomo 18q12.2-12.3.
OBJETIVOS
Estudo genético clínico da atrofia óptica autossômica dominante em 3 famílias.
Observar se o gene responsável está ligado aos marcadores polimórficos da região 3q28-29.
Verificar se há heterogeneidade genética, testando marcadores para o cromossomo 18q12.2-12.3.
MÉTODOS
Foram estudadas três famílias, denominadas A,B e C acometidas por Atrofia óptica autossômica dominante. Todos os membros destas famílias foram submetidos a exame oftalmológico que incluiu: acuidade visual corrigida, reflexos pupilares, oftalmoscopia binocular indireta sob midríase, com documentação fotográfica dos discos ópticos, campo visual, testes de visão de cores com placas pseudo-isocromáticas de Ishiraha (ISH) ou Hardy-Rand-Ritter (HRR). Nos pacientes com algum tipo de alteração sistêmica foram realizadas avaliações clínica e neurológica. Na ausência de outras alterações oculares, os pacientes com palidez do disco óptico foram classificados como afetados. Após a identificação do estado de afecção dos indivíduos, foram construídos heredogramas das famílias.
Esses indivíduos ou seus responsáveis foram informados sobre o estudo genético a ser realizado, quando da sua inclusão no protocolo. Foram assinados termos de consentimento informado para participação deste estudo.
O DNA genômico foi extraído das amostras de sangue periférico por tratamento com proteinase K e purificado pelo método de desalinização das proteínas celulares, desidratação e precipitação com uma solução saturada de NaCl e etanol(20).
A análise de ligação com dois pontos foi realizada com um painel de 11 marcadores fluorescentes polimórficos, tipo microssatélites, para a região 3q27-qter e 13 marcadores da região 18p11.2-18qter(20-22).
Marcadores cromossômicos foram obtidos da Research Genetics Inc. (MapPairsTM, Huntsville, Alabama, USA). Estes marcadores foram utilizados como oligonucleotídeos ("primers") nas reações de polimerase em cadeia (PCR) para amplificar regiões do DNA genômico que contém polimorfismos do tipo repetição de di, tri ou tetra nucleotídeos. As reações de PCR foram realizadas, utilizando-se 100 ng de DNA genômico, tampão 10 X (Perkin Elmer Cetus, EUA: 10 mM Tris pH 8,0, 0,01% gelatin, 50 mM KCI, 1,5 mM MgCl2), 125 mM de cada dNTP, 0,625 UI Taq polymerase (Perkin Elmer Cetus, EUA), 2,0 mM de cada oligonucleotídeo e água destilada estéril com um volume total de 12,5 ml. Após a desnaturação inicial procederam-se a amplificação em 30 ciclos de desnaturação, anelamento, otimização e extensão, em temperaturas específicas, na termocicladora (Perkin Elmer Cetus, EUA)(16).
Os produtos das reações de PCR foram fragmentos que continham as áreas de polimorfismos de diferentes tamanhos. A observação das variações destes alelos se fez pela identificação do peso molecular dos fragmentos. Para tanto, os produtos de PCR foram aplicados em gel de eletroforese de poliacrilamida 6% Gel-Mixâ6 (GibcoBRL, Life Technologiesä, EUA) em um seqüenciador (373 DNA sequencer ¾ Applied Biosystemsâ, EUA). Os tamanhos dos fragmentos foram identificados pelos programas "Genescam" e "Genotyper" (Applied Biosystemsâ, EUA)(16).
O peso molecular dos fragmentos foi utilizado como alelo nos heredogramas para análise da co-segregação. O programa "Cyriliic" (Cherwell Scientific, Inglaterra) foi utilizado para construção dos haplótipos dos indivíduos. A análise de ligação com dois pontos foi realizada usando-se o programa "Mink" (Columbia University, New York, NY, EUA)(23).
RESULTADOS
Cinqüenta e sete indivíduos das três famílias, A, B e C, foram submetidos ao exame oftalmológico para determinar o seu estado de afecção. Foram identificados 22 indivíduos afetados: 5 da família A, 10 da família B e 7 da família C. Dentre os afetados, 11 eram do sexo masculino e 11 do sexo feminino. As idades dos afetados variaram entre 5 e 81 anos. As acuidades visuais dos olhos afetados variaram entre 20/400 e 20/20. Todos os afetados apresentavam discromatopsia e palidez de disco óptico bilateral. Todos os indivíduos eram da raça branca, sendo uma das famílias brasileira e as demais americanas.
A atrofia óptica nestas famílias seguia o padrão de herança autossômico dominante com expressividade variável. A-l.1 e A-ll.1 apresentavam uma expressividade menor com alterações leves e eram assintomáticos.
A seguir encontram-se os heredogramas das três famílias. Os três primeiros heredogramas mostram os haplótipos dos indivíduos para os marcadores polimórficos do cromossomo 3 (Figuras 1, 2 e 3); e o último mostra os haplótipos dos indivíduos da família C para os marcadores polimórficos do cromossomo 18. (Figura 4)
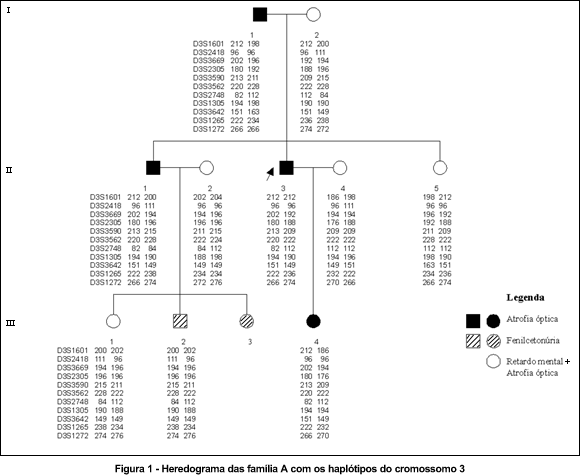
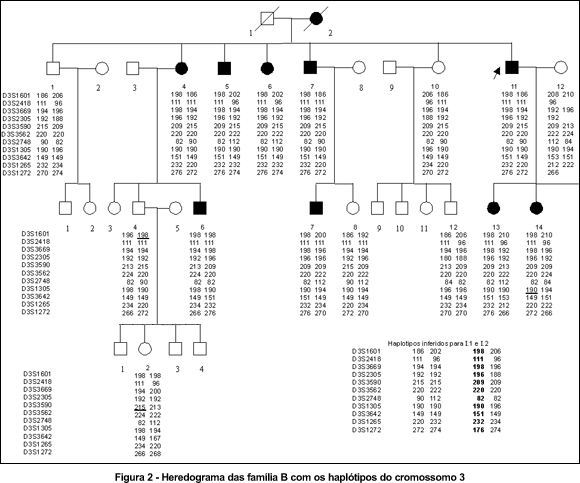
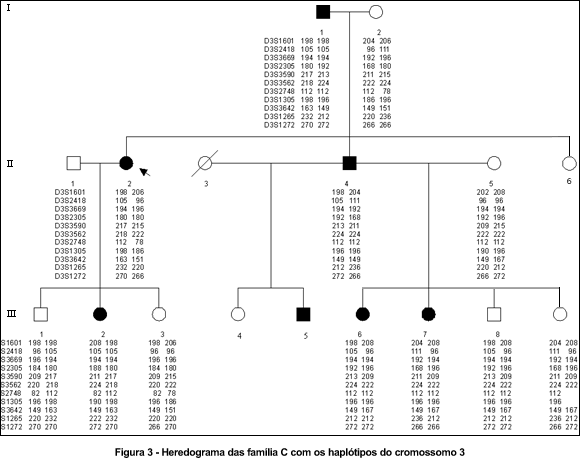
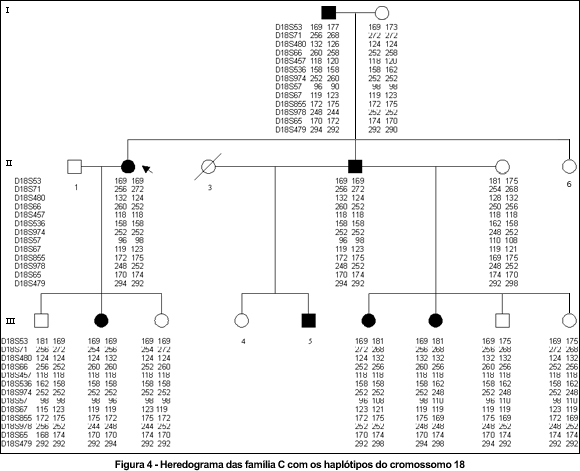
Os haplótipos de III.14 e IV.2 apresentam linhas que indicam o intervalo entre os marcadores onde houve evento de recombinação.
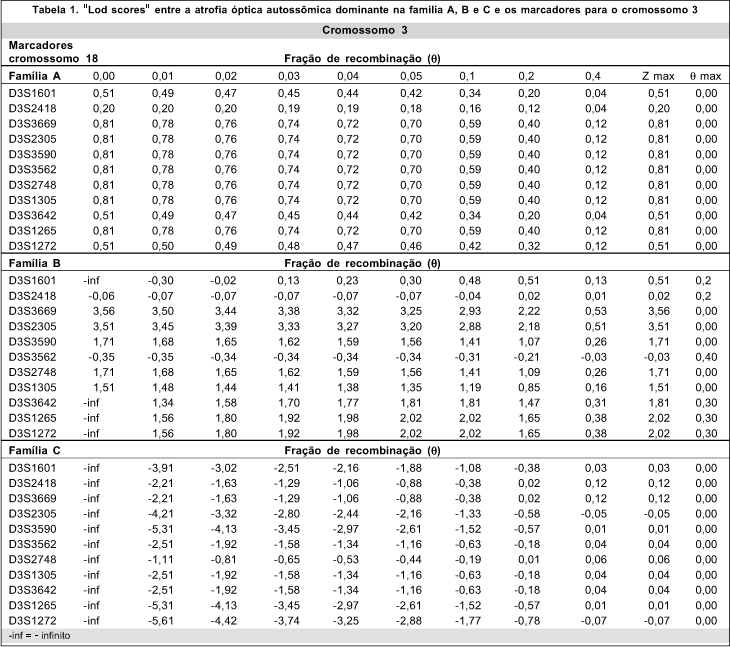
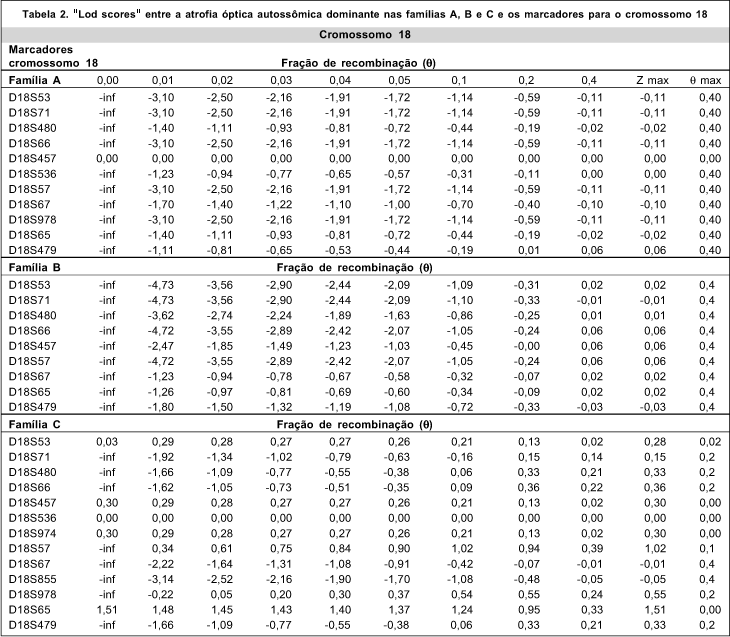
Para cada heredograma foi montada uma tabela com os valores de "lod scores" de dois pontos, isto é, entre cada marcador e a atrofia óptica (Tabelas 1 e 2).
Como as famílias A e B apresentaram ligação para o cromossomo 3 e não apresentavam para o 18 os heredogramas do cromossomo 18 não foram aqui inseridos. Entretanto das tabelas de "lod scores" apresentam os valores para estes heredogramas também.
DISCUSSÃO
A atrofia óptica autossômica dominante foi clinicamente semelhante nas três famílias, não sendo possível a distinção clínica mesmo após se conhecer que os genes mapeavam para loci diferentes.
Na análise da segregação dos haplótipos na família A para os marcadores do cromossomo 3, parece ter havido um haplótipo relacionado à atrofia óptica autossômica dominante. O "lod score" máximo obtido foi de 0,81 com fator de recombinação (0) de zero com os marcadores D3S3669, D3S2305, D3S3590, D3S2748, D3S1305, D3S3562 e D3S1265. Entretanto o "lod score" não alcançou o limite de 3,00 para que fosse estabelecida a significância estatística da ligação desta família no cromossomo 3q28-29 (Tabela 1). Isso porque se tratava de uma família pequena, com seis eventos de recombinação estudados, onde não é possível se atingir um "lod score" significante.
A família B mapeou para o cromossomo 3 com significância estatística, com "lod score" máximo de 3,56, fator de recombinação (0) de zero para o marcador D3S3669 no locus 3q28-29. (Tabela 1)
A região demarcada proximalmente pelo marcador D3S2418 e distalmente pelo D3S1305 mostrou-se ligada à doença sem nenhum evento de recombinação. Os marcadores incluídos nesta região eram: D3S2418, D3S3669, D3S2305, D3S3590, D3S3562, D3S2748 e D3S1305.
Dois eventos de recombinação nesta família situam o gene neste intervalo entre os marcadores D3S2418 e D3S1305. Houve recombinação entre os marcadores polimórficos em alguns indivíduos afetados. Observando-se o quadro 8 dos haplótipos das pacientes B-III.13, B-III.14 e B-III.4 é fácil notar que a paciente B-III.14 perdeu parte do haplótipo relacionado à doença num evento de recombinação ("crossing-over") distal entre D3S2748 e D3S3642 e o indivíduo normal B-III.4 herdou parte do haplótipo relacionado à doença, com um evento de recombinação proximal entre D3S1601 e D3S2418. (Quadro 1)
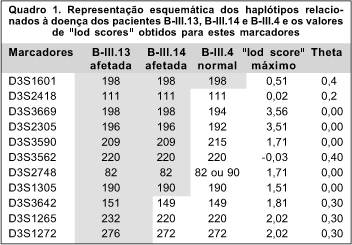
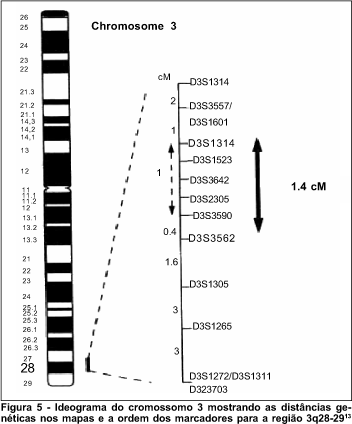
É difícil afirmar se este trabalho diminuiu a área de ligação pois, dependendo do mapa utilizado(4,21-24), a medida em centomorgans e a posição relativa dos marcadores variam. Analisando-se os marcadores que apresentaram ligação em outros trabalhos, conclui-se que a área de ligação neste trabalho é quase igual à encontrada por Brown e colaboradores e por Jonasdottir e cols que mediram 3,8 e 1,4 cM respectivamente(25,12). (Figura 5)
Foi excluída a ligação da atrofia óptica na família C com os marcadores situados na região cromossômica 3q (Tabela 1). Foi testada a ligação com marcadores do cromossomo 18, entretanto também não foi encontrada ligação. (Tabela 2)
A ausência de ligação dos loci 3q28-29 e 18q12.2-12.3 com a atrofia óptica na família C sugere a existência de um terceiro locus para a atrofia óptica. O que confirma que a atrofia óptica autossômica dominante apresenta heterogeneidade genética não alélica, isto é, nem todas as famílias mapeiam para o mesmo locus e provavelmente existe mais de um gene responsável pela atrofia óptica autossômica dominante. O gene OPA1 que se localiza em 3q28-29 parece ser o gene mais freqüentemente relacionado a esta doença. É possível que um segundo gene esteja localizado no cromossomo 18q12.2-12.3.
O refinamento atual do locus da atrofia óptica já permite a clonagem posicional deste gene, o que possibilitará identificação de genes candidatos na região crítica. O isolamento do gene humano para atrofia óptica não só melhorará nosso entendimento da fisiopatologia desta afecção ocular, mas também esclarecerá os mecanismos de diferenciação das células ganglionares e do desenvolvimento da retina humana.
CONCLUSÃO
O fato da terceira família não mapear para nenhum dos dois loci já descritos é indicativo de que existe heterogeneidade genética na atrofia óptica autossômica dominante e levanta a possibilidade de existir um terceiro locus para esta doença.
REFERÊNCIAS
1. Kjer P. Infantile optic atrophy with dominant mode of inheritance. A clinical and genetic study of 19 Danish families. Acta Ophthalmol (Copenh.) 1959;37(54 Suppl):1-146.
2. Hoyt CS. Autosomal dominant optic atrophy. A spectrum of disability. Oph-thalmology 1980;87:245-51.
3. Votruba M, Fitzke FW, Holder GE, Carter A, Bhattacharya SS, Moore AT. Clinical features in affected individuals from 21 pedigrees with dominat optic atrophy. . Arch Ophthalmol 1998;116:351-8.
4. Omim - Online Mendelian Inheritance in Man. Baltimore, MD., National Center for Biotechnology Information. . Available from URL: http://www.ncbi.nlm.nih.gov/omim
5. Smith DP. Diagnostic criteria in dominantly inherited juvenile optic atrophy. A report of three new families. Am J Optom Arch Am Acad Optom 1972; 49:183-200.
6. Johnston RL, Seller MJ, Behnam JT, Burdon MA, Spalton DJ. Dominant optic atrophy. Refining the clinical diagnostic criteria in light of genetic linkage studies. Ophthalmology 1999;106:123-8.
7. Kjer P, Jensen OA, Klinken L. Histopathology of eye, optic nerve and brain in a case of dominant optic atrophy. Acta Ophthalmol (Copenh.)1983;61:300-12.
8. KJer B, Eiberg H, Kjer P, Rosenberg T. Dominant optic atrophy mapped to chromossome 3q region II. Clinical and epidemiological aspects. Acta Ophthalmol Scand 1996;74:3-7.
9. Kivlin JD, Lovrien EW, Bishop DT, Maumenee IH. Linkage analysis in dominant optic atrophy. Am J Hum Genet 1983;35:1190-5.
10. Geitvik GA, Hoyheim B, Gedde-dahl T, Grzeschik KH, Lothe R, Tometer H, et al. The Kidd (JK) blood group locus assigned to chromosome 18 by close linkage to a DNA-RFLP. Hum Genet 1987;77:205-9.
11. Eiberg H, Kjer B, Kjer P, Rosenberg T. Dominant optic atrophy (OPA1) mapped to chromossome 3q region.I. Linkage analysis. Hum Mol Genet 1994;3:977-80.
12. Jonasdottir A, Eiberg H, Kjer B, Kjer P, Rosenberg T. Refinement of the dominant optic atrophy locus (OPA1) to a 1,4 cM interval on chromosome 3q28-3q29, within a 3-Mb YAC contig. Hum Genet 1997;99:115-20.
13. Votruba M, Moore A, Bhattacharya SS. Demonstration of a founder effect and fine mapping of a dominant optic atrophy locus on 3q28-qter by linkage disequilibrium method: a study of 38 British Isles pedigrees. Hum Genet 1998;102:79-86.
14. Bonneau D, Souied E, Gerber S, Rozet JM, D`Haens E, Journel H, et al. No evidence of genetic heterogeneity in dominant optic atrophy. J Clin Genet 1995;32:951-3.
15. Seller MJ, Behnam JT, Lewis CM, Johnston RL, Burdon MA, Spalton DJ. Linkage studies in a dominant optic atrophy, Kjer type: possible evidence for heterogeneity. J Med Genet 1997;34:967-72.
16. Kerrison JB, Arnould V, Stefko T, Barmada MM, Sallum JMF, VagefI R, et al. Autossomal dominant optic atrophy pedigree linked to chromosome 18q12.2-12.3 . Invest Ophthalmol Vis Sci 1998;39:2338.
17. The human genome: a guide to online information resources. Bethesda, M.D.: ational Center for Biotechnology Information. Available from the URL: http://www.ncbi.nlm.nih.gov/genome/guide.
18. Votruba M, Payne A, Moore AT, Bhattacharya SS. Dominant optic atrophy: exclusion and fine genetic mapping of the candidate gene HRY. Mamm. Genome 1998;9:784-7.
19. Stohr H, Klein J, Gehring A, Koehler MR, Jurklies B, Kellner U, et al. Mapping and genomic characterization of the gene encoding diacylglycerol kinase gamma (DAGK3): assessment of its role in dominant optic atrophy (OPA1). Hum Genet 1999;104:99-105.
20. Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2.ed. New York: Cold Spring Harbor Laboratory Press;1988.
21. GDB. - Genome Database. An international collaboration in support for the human genome Project. Toronto, Canadá. . Available from URL: http://www.gdb.org/gdb
22. LDB ¾ The genetic Location Database. Summary Maps. University of Southampton School of Medicine . Available from URL: http://cedar.genetics.soton.ac.uk/public_html/summarym.html
23. Terwillinger JT, Ott J. Handbook of human genetic linkage. Baltimore: Johns Hopkins University Press; 1994. 307p.
24. NCBI - National Center for Biotechnology Information. A new gene map of the human Genome. The International RH Mapping Consortium. GENEMAP`99. . Available from URL: http://www.ncbi.nlm.nih.gov/genemap99
25. Brown JR J, Fingert JH, Taylor CM, Lake M, Sheffield VC, Stone EM. Clinical and genetic analysis of a family affected with dominant optic atrophy (OPA1) . Arch Ophthalmol 1997;115:95-9.
NOTA DO AUTOR
Enquanto este trabalho aguardava publicação o gene OPA1 (OMIM*605290) foi identificado simultaneamente por Delettre e cols. (2000) e Alexander e cols. (2000)(1,2) no locus 3q28-q29. Este gene codifica um polipeptideo com homologia para uma "dynamin-related GTPases". Contém 28 exons que se localizam em 40kb de seqüência genômica. Existem 8 isoformas de RNAm dependendo da leitura dos exons 2 e 4. Mais de nove mutações já foram identificadas em diferentes famílias com OPA1(1-5).
As características patogênicas da OPA1 se assemelham as da Neuropatia Óptica de Leber, que resulta de um defeito na mitocôndria. Mutação em um gene do DNA mitocondrial acarreta diminuição no suprimento de energia nos neurônios do nervo óptico que consomem alta energia, principalmente do feixe papilo-macular. O transporte axonal das células ganglionares da retina é comprometido causando cegueira. Delettre 2001 sugere que mutações no gene OPA1 afetem a integridade mitocondrial, resultando em um déficit na produção energética.
REFERÊNCIAS
1. Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer PP, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 2000;26:207-10.
2. Alexander C, Votruba M, Pesch UEA, Thilselton DL, Mayer S, Moore A, et al. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominat optic atrophy linked to chromosome 3q28. Nat Genet 2000;26:211-5.
3. Pesch UEA, Leo-Kottler B, Mayer S, Jurklies B, Kellner U, Apfelstedt-Sylla E, et al. OPA1 mutations in patients with autosomal dominant optic atrophy and evidence for semidominant inheritance. Hum Mol Genet 2001;10:1359-68.
4. Toomes C, Marchbank NJ, Mackey DA, Craig JE, Newbury-Ecob RA, Bennett CP, et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum Mol Genet 2001;10:1369-78.
5. Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet 2001;109:584-91.
Parte da tese apresentada à Universidade Federal de São Paulo ¾ Escola Paulista de Medicina para obtenção do Título de Doutor em Medicina.
Este estudo foi realizado em parte graças ao suporte financeiro do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brasília, DF, Brasil.
1 Departamento de Oftalmologia da Universidade Federal de São Paulo (UNIFESP) São Paulo.
2 Johns Hopkins Center for Hereditary Eye Diseases ¾ Wilmer Eye Institute ¾ Johns Hopkins Hospital ¾ Baltimore, MD, USA.
Endereço para correspondência: Av. Indianópolis, 1797 ¾ São Paulo (SP) CEP 04063-003.
E-mail: [email protected]
Recebido para publicação em 28.08.2000
Aceito para publicação em 23.04.2002

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}