Arq. Bras. Oftalmol. 2002; 65 (6): 10.1590/S0004-27492002000600005
Total: 2772
Juliana Maria Ferraz Sallum1; Jane Chen2; Ana Beatriz Alvarez Perez
DOI: 10.1590/S0004-27492002000600005
RESUMO
Objetivo: Para se identificar as alterações oculares presentes na síndrome de Marfan, este trabalho apresenta a avaliação oftalmológica de 46 indivíduos portadores desta doença. Métodos: Estudo prospectivo com avaliação clínica de 46 pacientes portadores de síndrome de Marfan com avaliação oftalmológica completa. Dezessete pacientes também foram submetidos a exame genético clínico e estudo molecular. Resultados: Dos quarenta e seis pacientes incluídos neste estudo, as seguintes alterações oculares foram encontradas com maior freqüência: subluxação do cristalino (67,3%), hipoplasia de íris (23,9%), descolamento de retina (7,6%), córnea plana (2,2%), megalocórnea (2,2%) e miopia ou astigmatismo miópico (34,8%). Cinco pacientes (10,9%) apresentaram exame ocular normal em ambos os olhos. Detectou-se uma mutação patogênica distinta das relatadas na literatura em uma paciente, uma mutação de sentido trocado que ocorreu no éxon 28 levando à mudança de aminoácido C1166Y. Conclusões: As alterações oculares da síndrome de Marfan são freqüentes e o conhecimento do gene responsável FBN-1 e de sua expressão no olho auxiliam para o diagnóstico e tratamento destas anomalias.
Descritores: Síndrome de Marfan; Anomalias do olho; Oftalmopatias
ABSTRACT
Purpose: To identify the ocular abnormalities in Marfan´s syndrome patients. Methods: Prospective study of 46 Marfan patients with complete ophthalmologic evaluation. Seventeen patients also underwent a clinical genetic and molecular analysis. Results: Among the 46 patients included in this study, the following ocular abnormalities were found more frequently: ectopia lentis (67.3%), hypoplastic irides (67.3%), retinal detachment (7.6%), flattened cornea (2.2%), megalocornea (2.2%) and myopia (34.8%). Five patients (10.9%) presented normal ocular evaluation in both eyes. A pathogenic mutation different from those published in other studies was found in one patient. Conclusions: Ocular abnormalities in Marfan´s syndrome are frequent. The better understanding of the FBN-1 gene and its expression in the eye are very helpful tools for the diagnosis and treatment of the abnormalitites.
Keywords: Marfan's syndrome; Eye abnormalities; Eye diseases
INTRODUÇÃO
A Síndrome de Marfan (OMIM #154700) é uma doença do tecido conjuntivo cujas principais manifestações fenotípicas envolvem os sistemas esquelético, cardiovascular e ocular(1). Trata-se de uma doença de herança autossômica dominante que apresenta expressividade variável e penetrância completa. Baseados em estudos bioquímicos e genéticos, hoje se considera o gene FBN-1 (fibrilina-1) (OMIM *134797) como o responsável pela Síndrome de Marfan(1). O gene FBN-1 foi localizado no cromossomo 15q21 e é composto de 65 exons.
As principais manifestações esqueléticas são: deformidades do tórax e da coluna, dolicostenomelia, aracnodactilia e estatura elevada. As principais manifestações oculares são: subluxação do cristalino, miopia e descolamento de retina. As principais manifestações cardíacas são: prolapso de válvula mitral, dilatação da aorta e aneurisma dissecante de aorta. Podem ocorrer também manifestações no sistema nervoso central, como ectasia dural, meningocele lombar e sacral, cisterna magna dilatada, distúrbio de aprendizado e hiperatividade.
Dentre os sinais clínicos da Síndrome de Marfan alguns são considerados sinais maiores por apresentarem maior freqüência e especificidade, são eles: subluxação do cristalino, dilatação da aorta ascendente, dissecção da aorta e ectasia dural. Muitas destas manifestações são idade dependentes, como a dilatação da aorta; e outras são de difícil investigação, como a ectasia dural(2). A Síndrome de Marfan apresenta grande variabilidade fenotípica inter e intrafamilial, o que torna o diagnóstico difícil.
Os critérios atuais estabelecidos na literatura para:
1) diagnóstico do primeiro afetado da família são:
- se a história familial/genética não contribui: critério maior em pelo menos dois diferentes órgãos/sistemas e o envolvimento de um terceiro;
- se uma mutação conhecida como causadora da Síndrome de Marfan for detectada: um critério maior em um órgão/sistema e o envolvimento de um segundo.2) um parente de um caso índice: presença de um critério maior na história familial e um critério maior em um órgão/sistema e o envolvimento de um segundo.
Hoje se sabe que o diagnóstico de Síndrome de Marfan deve ser considerado mesmo se o paciente não cumpra os critérios diagnósticos, desde que seja criteriosamente investigado. São chamados de Síndrome de Marfan os pacientes que cumprem os critérios diagnósticos. Além disso, hoje existe um diagnóstico mais amplo chamado de fibrilinopatias.
Fazendo parte da variação fenotípica desta doença existe um quadro chamado de Síndrome de Marfan Neonatal (OMIM #154700) cujo quadro clínico é considerado mais grave. Pode estar presente ao nascimento ou muito precocemente(1). Aproximadamente 85% dos casos são esporádicos. É caracterizada por alterações cardíacas graves, contraturas congênitas, aracnodactilia, dolicocefalia, palato alto e arqueado, micrognatia, hipermobilidade articular, pés planos, deformidade anterior do tórax, iridodonese, megalocórnea e luxação do cristalino. As mutações que levam ao quadro de SMN tendem a se concentrar em uma região central do gene FBN-1 codificada pelos éxons 24-32(2).
Os principais diagnósticos diferenciais da Síndrome de Marfan a serem considerados incluem as doenças que apresentam a manifestação ocular mais notável desta síndrome, a subluxação do cristalino. São elas: síndrome de Weill-Marchesani e homocistinúria. O principal diagnóstico diferencial da Síndrome de Marfan Neonatal (SMN) deve ser feito com a aracnodactilia contratural congênita (ACC).
A Síndrome de Weill-Marchesani (OMIM *277600) é uma doença de herança autossômica mais freqüentemente recessiva. É caracterizada por subluxação do cristalino mais comumente inferior, baixa estatura e braquidactilia (dedos largos e curtos)(1). Outra manifestação ocular é a microesferofacia, que pode levar à miopia(3). Nesta doença é freqüente que o cristalino microesferofácico luxe para a câmara anterior, causando um glaucoma agudo.
A homocistinúria (OMIM *236200) é um erro inato do metabolismo(1). É causado principalmente por deficiência da cistationina beta-sintetase levando à produção aumentada de homocisteína e metionina na urina, podendo ser também a manifestação de outras deficiências de enzimas do metabolismo da cisteína. É uma condição de herança autossômica recessiva e as principais manifestações clínicas envolvem os sistemas nervoso central, ocular, músculo-esquelético e vascular. Os pacientes podem apresentar retardo mental e desordens psiquiátricas. As manifestações oculares e esqueléticas são sugestivas de síndrome de Marfan exceto por apresentarem subluxação do cristalino mais comumente para baixo, mobilidade articular limitada e osteoporose(3). Apresentam também fenômenos tromboembólicos e hipopigmentação, sendo que na maioria dos pacientes a íris é de cor azul. Em todos os casos suspeitos de Síndrome de Marfan, a homocistinúria deve ser excluída através da análise de aminoácidos na urina e sangue.
A aracnodactilia contratural congênita (OMIM *121050) é uma doença do tecido conjuntivo de herança autossômica dominante(1). O gene responsável é o fibrilina-2 (FBN-2), localizado no cromossomo 5q23-31. As similaridades entre a SMN e a ACC grave são: aracnodactilia, contraturas congênitas múltiplas e algumas características faciais. Ambas apresentam anormalidades cardiovasculares severas que levam à morte precoce, mas as alterações cardíacas específicas são diferentes: na SMN pode ocorrer insuficiência valvular e dilatação da aorta, e na ACC, defeitos estruturais, sendo o prolapso de válvula mitral a principal manifestação. Escoliose e anomalias vertebrais tem maior prevalência na ACC. Atresias de duodeno e de esôfago, e má rotação intestinal só ocorrem nesta entidade. A ACC apresenta também orelhas pregueadas e complicações oculares variadas, dentre elas a miopia e o ceratocone.
A fibrilina, proteína da matriz extracelular do tecido conjuntivo, é um componente da malha microfibrilar que se apresenta amplamente distribuído por todo o corpo. Na síndrome de Marfan, a fibrilina é estrutural e funcionalmente anormal dependendo do tipo de mutação(4).
O gene da fibrilina se expressa em grande parte do tecido conjuntivo ocular. A distribuição da fibrilina em tecidos oculares humanos normais foi investigada por técnicas de imunohistoquímica sendo encontrado uma ampla distribuição da mesma no tecido conjuntivo ocular(5). A fibrilina é fisiologicamente ausente no vítreo e na retina(5).
O gene da fibrilina-2 é homólogo ao da fibrilina-1, mutações nestes genes causam desordens fenotipicamente semelhantes. Alguns autores sugerem que a expressão do gene FBN-2 direciona a montagem das fibras elásticas durante a embriogênese e a do gene FBN-1 apresenta uma função estrutural maior das microfibrilas.
Os objetivos deste estudo são identificar as alterações oculares presentes em pacientes com a síndrome de Marfan, pesquisar mutações no gene da fibrilina, correlacionar as alterações oculares com as mutações encontradas e discutir o tratamento das alterações oculares.
MÉTODOS
Estudo prospectivo, com avaliação clínica e oftalmológica de quarenta e seis pacientes portadores de Síndrome de Marfan, realizado entre os anos de 1992 e 1999. A idade dos pacientes variou de 1 ano e 3 meses a 45 anos, sendo 31 do sexo feminino e 15 do sexo masculino.
Dezessete pacientes foram submetidos a exame genético clínico para avaliar a presença de alterações sistêmicas da Síndrome de Marfan.
A avaliação oftalmológica completa de indivíduos portadores da Síndrome de Marfan foi feita de acordo com a idade e colaboração do paciente. Quando possível, incluiu: história, medida da acuidade visual, refração, avaliação da musculatura ocular extrínseca, biomicroscopia, tonometria de aplanação e oftalmoscopia binocular indireta sob midríase; além de exames complementares para alguns pacientes, por exemplo, ultra-sonografia, ceratoscopia computadorizada.
O exame clínico-genético consta da propedêutica genético clínica com anamnese e exame físico completos e medidas antropométricas específicas, e a realização de exames especiais e complementares, dentre elas, eletrocardiograma, radiografia de tórax, ecocardiograma, mapeamento de fluxo, doppler pulsátil, radiografia de coluna, conforme a necessidade do caso.
Foram obtidos dados sobre os antecedentes familiares de todos pacientes, entretanto somente o primeiro afetado na família foi incluído no estudo clínico e molecular. Em todos os pacientes foi excluída a homocistinúria pelo teste urinário.
O estudo molecular realizado constou inicialmente da extração do DNA genômico a partir dos leucócitos do sangue periférico. Os 65 éxons do gene FBN-1 dos 17 pacientes foram amplificados pela técnica de PCR (Polymerase Chain Reaction ou Reação em Cadeia da Polimerase), tendo sido utilizados 65 pares de oligonucleotídeos. Os produtos da PCR foram submetidos à pesquisa das mutações pelos métodos de SSCP "Single Strand Conformation Polymorphism" ou, em português, Polimorfismo de Conformação de Fita Simples de DNA, e de heteroduplex. Quando um padrão alterado de bandas foi observado ao SSCP ou heteroduplex, foi realizado o seqüênciamento do produto correspondente ao éxon.
RESULTADOS
Dos quarenta e seis pacientes incluídos neste estudo, cinco pacientes (10,9%) apresentaram exame ocular normal em ambos os olhos (Tabela 1).
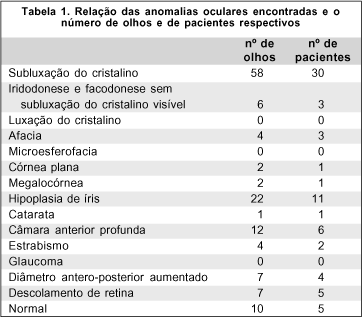
A subluxação do cristalino foi observada em cinqüenta e oito olhos de trinta pacientes (63,0%). Destes, dois pacientes apresentaram subluxação do cristalino em um olho e afacia no outro olho. Outro paciente apresentava afacia nos dois olhos. A afacia nestes pacientes era pós-cirurgia para tratamento da subluxação. O que sugere que a real freqüência de subluxação seja 67,3%.
Três pacientes (6,5%) apresentaram iridodonese e facodonese na ausência de subluxação do cristalino visível ao exame. Nenhum paciente apresentou luxação do cristalino para a câmara anterior ou para o vítreo.
A microesferofacia não foi observada em nenhum paciente. Um paciente (2,2%) apresentou córnea plana ao exame e um paciente (2,2%) apresentou megalocórnea.
Foi observada hipoplasia de íris em onze pacientes (23,9%), com conseqüente dificuldade para dilatação da pupila. Catarata unilateral foi observada em um paciente, câmara anterior profunda em seis pacientes, estrabismo em dois pacientes, e não foi encontrado glaucoma em nenhum paciente.
Quatro pacientes (7,6%) apresentaram diâmetro ocular ântero-posterior aumentado, destes um apresentava um olho atrófico. Cinco pacientes (7,6%) apresentaram descolamento de retina, sendo que em dois era bilateral.
Dos quarenta e seis pacientes, vinte e quatro eram emétropes (52,1%) e em um não foi possível realizar a refração. Dezesseis pacientes (34,8%) apresentaram miopia ou astigmatismo miópico, sendo que em dois foi observada alta miopia. Seis pacientes (13,0%) apresentaram hipermetropia ou astigmatismo hipermetrópico (Tabela 2).

Nos 17 pacientes submetidos a exames genéticos detectou-se alteração no padrão de bandas do SSCP e/ou heteroduplex em um paciente, sendo realizado seqüenciamento para caracterizar a mutação. A mutação encontrada foi de sentido trocado (G®A em 3497) que ocorreu no éxon 28 levando à mudança de aminoácido C1166Y.
DISCUSSÃO
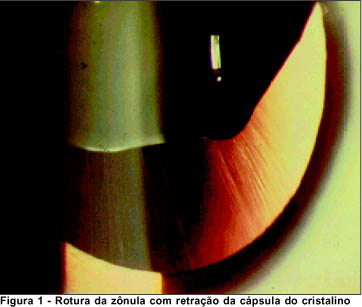
No olho normal as moléculas de fibrilina ficam dispostas como contas de colar sobre o colágeno(6). Na Síndrome de Marfan a fibrilina anormal acarreta deslizamento das bandas de colágeno, provocando alongamento da zônula. Este alongamento da zônula provoca retração da cápsula cristaliniana com alteração secundária da forma do cristalino. O cristalino torna-se mais globoso pela falta de tensão zonular sobre a cápsula. Além disso, existem menos processos ciliares, os quais apresentam orientação irregular e sinais de degeneração. Na cápsula do cristalino a fibrilina anormal, localizada na região equatorial, leva a alterações no desenvolvimento e funcionamento do cristalino(5) (Figura 1).
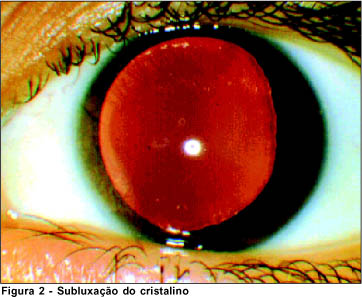
A subluxação do cristalino, também denominada "ectopia lentis", é a manifestação ocular mais comumente encontrada nesta síndrome. Refere-se ao deslocamento do cristalino da sua posição normal no eixo visual. Relatos da literatura demonstraram que 60 a 80% dos casos apresentam subluxação do cristalino(2,7). É usualmente bilateral e o cristalino pode estar deslocado em qualquer direção, sendo que em 77% dos casos encontra-se deslocado para cima, nasal superior ou temporal superior(2,7). A subluxação tem um caráter progressivo documentado em aproximadamente 7,5% dos casos de literatura(2,7). Provavelmente o caráter progressivo seja mais freqüente. A subluxação do cristalino manifesta-se por baixa acuidade visual, de intensidade variável, dependendo da intensidade da subluxação e da existência de outras alterações oculares associadas(3). A cápsula cristaliniana retrai nas áreas não tracionadas pela zônula. Como isto ocorre em geral na região nasal inferior, é comum que o cristalino fique com a margem retificada nesta região e deslocado no sentido contrário(6). Neste estudo, foi observado subluxação do cristalino em 67,3% dos casos, sendo bilateral em todos os casos. É opinião dos autores deste trabalho que talvez as fibras zonulares da região nasal inferior, correspondente à área embrionária de fechamento da vesícula óptica, sejam mais vulneráveis a alterações da fibrilina. Por isso poderiam romper mais freqüentemente e causar retração da cápsula cristaliniana nesta região. Com deslocamento do cristalino em sentido contrário, temporal superior (Figura 2).
A iridodonese e a facodonese são sinais sugestivos de subluxação do cristalino. A iridodonese decorre da falta de suporte para a íris. Neste estudo, foi observado iridodonese e facodonese em todos os olhos que apresentaram subluxação do cristalino e em 6,5% dos casos estavam presentes na ausência de subluxação do cristalino visível ao exame. É freqüente a iridodonese e a facodonese serem os primeiros sinais clínicos da distopia cristaliniana.
Quando o cristalino é deslocado totalmente, caracteriza-se a luxação do cristalino. A luxação para o espaço vítreo é rara e pode complicar-se em glaucoma facolítico(11). A luxação para a câmara anterior também é pouco freqüente e pode produzir glaucoma agudo por bloqueio pupilar. Não foi observado luxação do cristalino em nenhum paciente deste estudo. Sempre que ocorrer subluxação para a câmara anterior deve-se suspeitar que o diagnóstico não seja de Síndrome de Marfan e sim de homocistinúria ou Síndrome de Weill-Marchesani.
A microesferofacia é rara, mas pode ocorrer resultando em miopia. Ela ocorre quando há desprendimento da zônula em 360º reduzindo a tensão na cápsula do cristalino e provocando uma alteração da forma do cristalino. Ele torna-se mais globoso. Não foi observado microesferofacia em nenhum paciente deste estudo.
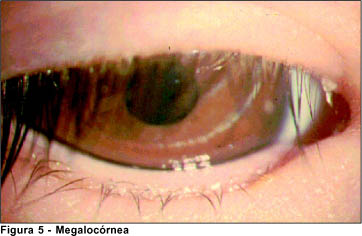
A córnea é plana em aproximadamente 15% dos casos(7). Megalocórnea é um achado comum. Foi observado córnea plana bilateral em um paciente e megalocórnea bilateral em um paciente. O paciente com megalocórnea era uma criança com Síndrome de Marfan Neonatal. A fibrilina está presente na membrana basal do epitélio e entre as lamelas do colágeno no estroma corneanos.
A íris é freqüentemente hipoplásica, apresentando-se com poucos relevos e pode transiluminar em 10% dos casos, quando há desgaste crônico por toque do cristalino no epitélio posterior da íris(7). A pupila pode ter forma anormal e miótica, e ser difícil de dilatar nos casos mais severos, o que torna o exame da retina e as cirurgias mais difíceis. Neste trabalho foi observado hipoplasia de íris em 23,9% dos casos. A causa desta dificuldade se deve ao fato de a fibrilina estar presente na matriz do tecido conjuntivo e em abundância na região dos músculos esfíncter e dilatador (Figura 3).

O estrabismo é mais prevalente em pacientes com Síndrome de Marfan do que na população geral(8). Foi realizado um estudo de 573 pacientes portadores de Síndrome de Marfan objetivando avaliar a ocorrência de estrabismo, erros de refração e ambliopia(8). Destes pacientes, 19,2% apresentaram estrabismo, sendo a exotropia (11,7%) o tipo mais comum, seguido da esotropia (2,1%). Foi encontrada ambliopia associada a estrabismo em 44,5% dos pacientes. No presente trabalho, o estrabismo estava presente em 2 pacientes.
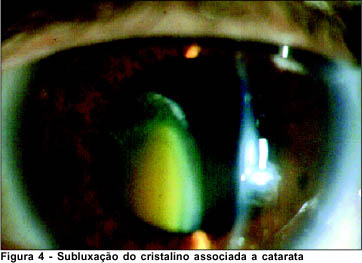
O cristalino subluxado desenvolve catarata com maior freqüência. A prevalência de todos os tipos de glaucoma é maior em pacientes com Síndrome de Marfan do que na população geral, sendo o glaucoma primário de ângulo aberto o tipo mais freqüente(9). A ocorrência de glaucoma é de aproximadamente 5%(9). Foi observado catarata unilateral em um paciente, e não foi observado glaucoma em nenhum paciente (Figura 4).
A fibrilina anormal leva a um retardo no desenvolvimento do corpo ciliar e alterações na estrutura do ângulo da câmara anterior, inclusive alterações no desenvolvimento do canal de Schlemm(5).
O comprimento axial do globo ocular aumenta progressivamente nestes pacientes, o que contribui para o aumento do componente axial da miopia, o descolamento de retina e a subluxação do cristalino. A biometria e o ultra-som são úteis para o diagnóstico e acompanhamento do crescimento do diâmetro ântero-posterior. Este aumento do globo ocular causa complicações vítreo-retinianas semelhantes às da alta miopia, rarefação do epitélio pigmentário retiniano e degenerações periféricas da retina. Foi medido o comprimento axial do globo ocular em quatro pacientes que apresentavam alterações fundoscópicas de alta miopia para acompanhamento deste crescimento ao longo do tempo. Talvez este exame seja útil também para programação cirúrgica ou para se avaliar prognóstico cirúrgico.
As ametropias mais encontradas são: miopia e astigmatismo de índice por alteração da conformação do cristalino, miopia e astigmatismo axiais por alteração do diâmetro antero-posterior do globo ocular. As alterações tipicamente encontradas na miopia axial são rarefação do epitélio pigmentário retiniano, coróide fina, crescentes esclerais peripapilares e estafiloma globoso do pólo posterior. Foi observado miopia ou astigmatismo miópico em 34,8% dos casos.
O descolamento de retina representa a complicação ocular mais grave desta síndrome. Foi encontrado em um estudo descolamento de retina em 16 olhos de 13 pacientes entre 160 pacientes com Síndrome de Marfan, sendo que em 3 pacientes foi bilateral(7). Ocorre com freqüência em olhos com miopia axial após extração do cristalino(6). Alterações periféricas da retina são comumente encontradas, principalmente degeneração branco sem pressão, degeneração lattice e roturas retinianas(7). O descolamento de retina foi encontrado em 5 pacientes (7,6%), sendo unilateral em três pacientes e bilateral em dois.
Um paciente com Síndrome de Marfan Neonatal foi avaliado e ao exame ocular notava-se megalocórnea em ambos os olhos (Figura 5).
A correlação entre o genótipo e o fenótipo observado nas diferentes mutações encontradas até o momento baseia-se nos relatos dos distintos quadros clínicos, no tipo da mutação ou nos grupos de mutações que podem ter um efeito semelhante e, sobretudo, nos achados imunohistoquímicos e nas análises da biossíntese da fibrilina neste grupo de mutações(2).
As mutações relatadas na literatura que apresentam fenótipo de Síndrome de Marfan predominantemente ocular são(2) as que ocorrem no éxon 21 que levam à mudança de aminoácido C890R e no éxon 59 com a mudança de aminoácido E2447K. A mutação encontrada em um paciente deste estudo foi de sentido trocado que ocorreu no éxon 28 levando à mudança de aminoácido C1166Y. A paciente é uma menina de 8 anos e nove meses, caso esporádico, filha de casal não consangüíneo. Apresentava subluxação do cristalino bilateral e alta miopia além de outras manifestações não oculares, correspondendo ao fenótipo de Síndrome de Marfan clássica.
O tratamento das alterações oculares se baseia no acompanhamento para diagnóstico precoce das complicações da subluxação do cristalino, glaucoma, e alterações vitreorretinianas, e correção óptica das ametropias. A prescrição de óculos pode ser difícil mas é a indicação na grande maioria dos casos. O uso de lentes de contato pode ser indicado para correção de afacia em alguns casos. A cirurgia para correção de subluxação com retirada do cristalino deve ser indicada apenas quando a correção óptica não for satisfatória, pois a incidência de complicações, como glaucoma e descolamento de retina, é alta. A técnica cirúrgica varia muito desde facoemulsificação à extração extra ou intracapsular, ou ainda lensectomia dependendo da localização e grau de subluxação do cristalino. Na opinião dos autores deste trabalho, a incidência de complicações parece ser mais freqüente nos casos com diâmetro antero-posterior do globo ocular aumentado.
CONCLUSÕES
As alterações oculares da Síndrome de Marfan são freqüentes e o conhecimento do gene responsável FBN-1 e de sua expressão no olho auxiliam o diagnóstico e tratamento destas anomalias. Este trabalho identificou uma mutação no gene da FBN-1 distinta das relatadas na literatura, uma mutação de sentido trocado que ocorreu no éxon 28 levando à mudança de aminoácido C1166Y. A correlação clínico-molecular ainda é difícil pois mutações no gene FBN-1 nem sempre são encontradas. Talvez porque as mutações não estejam nas áreas codificadas do gene ou porque existam outros genes responsáveis.
COMENTÁRIOS
Hoje em dia existe uma tendência de que estes indivíduos sejam acompanhados por um grupo multiprofissional devido às alterações presentes em muitos órgãos. É importante que existam centros de referência para o tratamento dos olhos destes pacientes pois as complicações são freqüentes e necessitam de oftalmologistas com experiência.
REFERÊNCIAS
1. Online Mendelian Inheritance in Man, OMIM (TM) . Baltimore: Johns Hopkins University; 2001. . Avaliable from:URL: http://www.ncbi.nlm.nih.gov/omim/.
2. Perez ABA. Estudo genético-clínico e molecular em pacientes com síndrome de Marfan . São Paulo: Universidade Federal de São Paulo; 1997.
3. Nelson LB, Maumenee IH. Ectopia lentis. Surv Ophthalmol 1982;27:143-60.
4. Kielty CM, Davies SJ, Phillips JE, Jones CJ, Shuttleworth CA, Charles SJ. Marfan syndrome: fibrillin expression and microfibrillar abnormalities in a family with predominant ocular defects. J Med Genet 1995;32:1-6.
5. Wheatley HM, Traboulsi EI, Flowers BE, Maumenee IH, Azar D, Pyeritz RE, Whittum-Hudson JA. Immunohistochemical localization of fibrillin in human ocular tissues ¾ Relevance to the Marfan syndrome. Arch Ophthalmol 1995;113:103-9.
6. Traboulsi EI: Ectopia lentis and associated systemic disease. In: Wiggs J, editor. Molecular genetics of ocular disease. New York: Wiley-Liss; 1995, p.219-33.
7. Maumenee IH. The eye in Marfan syndrome. Trans Am Ophthalmol Soc 1981;79:684-733.
8. Izquierdo NJ, Traboulsi EI, Enger C, Maumenee IH. Strabismus in the Marfan syndrome. Am J Ophthalmol 1994;117:632-5
9. Izquierdo NJ, Traboulsi EI, Enger C, Maumenee IH. Glaucoma in the Marfan syndrome. Trans Am Ophthalmol Soc 1992;90:111-7.
1 Chefe do Setor de Retina e Vítreo do Departamento de Oftalmologia da Universidade Federal de São Paulo - UNIFESP. Realizou todos os exames oftalmológicos.
2 Aluna de Medicina da Universidade Federal de São Paulo - UNIFESP.
3 Médica contratada, Doutora em Morfologia pela Universidade Federal de São Paulo - UNIFESP.
Endereço para correspondência: Av. Indianópolis, 1797 - São Paulo (SP) CEP 04063-003.
E-mail: [email protected]
Recebido para publicação em 14.11.2001
Aceito para publicação em 04.03.2002
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. José Carlos Eudes Carani.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.