Arq. Bras. Oftalmol. 2004; 67 (1): 10.1590/S0004-27492004000100026
Total: 2591
Patrick Frensel de Moraes Tzelikis1; Luciene Chaves Fernandes2
DOI: 10.1590/S0004-27492004000100026
RESUMO
OBJETIVO: Avaliar e comparar com a literatura o perfil dos pacientes portadores de colobomas oculares, bem como verificar a acuidade visual, alterações oculares associadas e alterações sistêmicas apresentadas pelos mesmos. MÉTODOS: Foram estudados retrospectivamente 18 pacientes com diagnóstico de coloboma ocular matriculados no serviço de Visão Subnormal do Hospital São Geraldo da Universidade Federal de Minas Gerais (HSG - HCUFMG). Foram avaliados: sexo, idade, raça, história familiar de anomalias oculares, história gestacional, tipo de coloboma, localização, bilateralidade, presença de microftalmia ou anoftalmia, alterações oculares concomitantes, e associação com doenças sistêmicas e acuidade visual. RESULTADO: Dez (55,6%) pacientes eram do sexo feminino, 15 (83,3%) eram leucodérmicos. A idade variou de 4 a 57 anos, com média de 9,5 anos. Apenas 3 (16,7%) apresentavam história familiar para coloboma. Todos apresentavam história gestacional negativa para qualquer intercorrência. Houve acometimento bilateral em 100% dos casos, com microftalmia presente em 6 (33,3%) casos. O coloboma foi típico em 14 (77,8%) pacientes, sendo o coloboma mais comum o retinocoroidal ocorrendo em 16 (88,9%) pacientes. 50% dos pacientes apresentavam o coloboma como achado isolado, e a outra metade associado a alguma doença sistêmica. Em 14 (77,8%) pacientes foi encontrado algum tipo de alteração ocular além do coloboma. CONCLUSÃO: O exame oftalmológico completo é muito importante tanto no diagnóstico quanto no prognóstico de pacientes com coloboma, visto que este pode estar associado a uma baixa visual importante, além de poder estar relacionados a diferentes anomalias ou síndromes.
Descritores: Coloboma; Baixa visão; Microftalmia
ABSTRACT
PURPOSE: To verify and compare with the literature the profile of patients with ocular coloboma, and also to verify visual acuity, ocular and systemic abnormalities. METHODS: Retrospective analysis of 18 patients with a definite diagnosis of ocular coloboma at the Low Vision Department of the São Geraldo Eye Hospital of the Federal University of Minas Gerais. Patients underwent a complete ophthalmologic evaluation: sex, age, race, family history, prenatal and birth history, complete or incomplete coloboma, location, microphthalmos or anophthalmos, other ocular and systemic abnormalities, and visual acuity. RESULTS: 10 (55.6%) patients were women. The age was from 4 to 57, and the mean was 9.5 years. Only three (16.7%) patients had a family history of coloboma. All patients had negative prenatal and birth history. The coloboma was bilateral in 100% of the patients, and associated with microphthalmos in 6 (33.3%) cases. The coloboma was considered typical in 14 (77.8%) patients, with retinochoroidal coloboma being the most frequent, 16 (88.9%) patients. 50% of the patients had coloboma as an isolated ocular anomaly, while the other 50% presented association with systemic anomalies. In 14 (77.8%) patients other ocular abnormalities were found. CONCLUSION: The complete ophthalmologic examination is extremely important for both the diagnosis and prognosis of the patients with coloboma, since the coloboma may be associated with important low visual and also systemic anomalies and syndromes.
Keywords: Coloboma; Vision, low; Microphthalmos
INTRODUÇÃO
Anomalias do desenvolvimento do globo ocular podem ocorrer em várias etapas da embriogênese. Coloboma ocular é uma destas malformações oculares relacionadas a um defeito na embriogênese(1).
Os colobomas podem acometer qualquer estrutura ocular, incluindo pálpebra, córnea, íris, zônula e corpo ciliar, coróide, retina e nervo óptico. As etiologias do coloboma são bastante variadas sendo a maioria de aparecimento isolado sem doença sistêmica associada.
Os colobomas podem ser típicos ou atípicos. Os colobomas típicos, mais comuns, seriam aqueles de localização nasal inferior, causados por um defeito no fechamento da fissura fetal(2). E os colobomas atípicos seriam todos aqueles de localização diferente da nasal inferior, de causa ainda não esclarecida(3).
Várias destas anomalias oculares podem cursar com visão subnormal. O intuito deste trabalho é estabelecer uma correlação entre os diferentes colobomas e a função visual apresentada pelo paciente além de mostrar a importância de um diagnóstico precoce para um melhor prognóstico visual.
MÉTODOS
Este trabalho foi realizado no Serviço de Visão Subnormal do Hospital São Geraldo da Universidade Federal de Minas Gerais (HSG - UFMG). Foram analisados retrospectivamente os prontuários de todos os pacientes com diagnóstico de coloboma ocular matriculados no Serviço de Visão Subnormal do HSG entre janeiro de 1992 e dezembro de 2001, totalizando 18 pacientes.
Foi avaliado a idade, sexo, raça, história familiar de anomalias oculares e história gestacional. Quanto ao exame oftalmológico foi anotada a acuidade visual obtida pelos testes do Olhar Preferencial (Teller Acuity Cards), tabela LH (Lea Hyvärinen) para os iletrados ou ETDRS (Early Treatment Diabetic Retinopathy Study) para os letrados. Para classificação das perdas visuais, considerou-se a Classificação de perdas visuais da Organização Mundial de Saúde e Conselho Internacional de Oftalmologia (CID-9-CM) que considera: Visão normal ou quase: AV de 20/12 a 20/60; Visão subnormal moderada: AV de 20/80 a 20/160; severa: AV de 20/200 a 20/400 e visão subnormal profunda: AV de 20/500 a 20/1000; cegueira ou quase: AV de 20/1250 a ausência de percepção de luz. Verificou-se as estruturas oculares envolvidas no coloboma, sua lateralidade e localização, se típica ou atípica, segundo as anotações da biomicroscopia e fundoscopia existentes no prontuário. Registrou-se a presença de microftalmia ou anoftalmia através de dados de ultra-sonografia. Pesquisou-se alterações sistêmicas associadas e outras alterações oculares concomitantes. Todos os dados foram anotados em um protocolo específico e então avaliados.
RESULTADOS
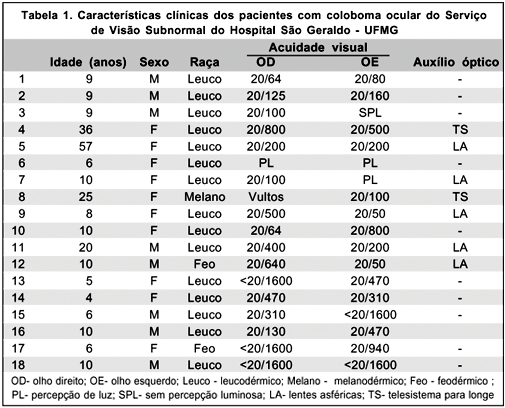
Foram encontrados 18 pacientes (0,75%) com coloboma ocular de um total de 1325 pacientes matriculados no Serviço de Visão Subnormal no período de janeiro de 1992 a dezembro de 2001. Destes, oito (44,4%) eram do sexo masculino, sendo 10 ( 55,6%) do sexo feminino. A idade variou de 4 a 57 anos, com média de 9,5 anos. Quanto à cor, 15 (83,3%) eram leucodérmicos, 2 (11,1%) feodérmicos e apenas um (5,6%) melanodérmico (Tabela 1).
Todos os pacientes apresentavam história gestacional negativa para qualquer intercorrência (infecções, inflamações, irradiações) ou uso de medicações. A grande maioria dos pacientes não apresentava nenhuma história familiar para coloboma ou qualquer outra patologia ocular, sendo que 3 (16,7%) pacientes eram irmãos. Não havia história de consangüinidade entre os pais dos 18 pacientes estudados.
A acuidade visual no olho direito variou de 0,014 (20/1400) a 0,3 (20/60 ) com média de 0,1 (20/200). No olho esquerdo a acuidade visual variou de ausência de percepção luminosa até 0,4 ( 20/50 ) com média também de 0,1 (20/200). A prescrição de auxílios ópticos foi realizada em sete casos (38,9%), sendo que a maioria dos pacientes (55,6%) necessitou apenas de orientação pessoal, familiar e/ou escolar.
Dos 18 pacientes com coloboma, 7 pacientes (38,9%) faziam uso de algum tipo de auxílio óptico, sendo que em 5 casos (27,7%) os pacientes utilizavam óculos com lentes asféricas para correção óptica para perto, e em 2 casos (11,2%) telesistemas monoculares para longe.
O acometimento foi bilateral em todos os pacientes estudados (100%), sendo que a microftalmia esteve presente em 6 (33,3%) e não houve caso de anoftalmia. O coloboma se apresentava típico, nasal inferior, em 14 pacientes (77,8%) e atípico em 4 (22,2%). Quanto à localização do coloboma 16 pacientes (88,9%) apresentavam coloboma retinocoroidal, 14 (77,8%) coloboma de nervo óptico, 10 (55,6%) coloboma de íris, 2 (11,1%) coloboma de mácula e apenas 1 (5,6%) coloboma de zônula e corpo ciliar (Tabela 2).

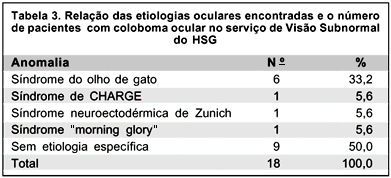
Em relação à etiologia 9 (50,0%) pacientes não apresentaram nenhuma causa definida ou doença associada, 6 (33,2%) apresentavam diagnóstico de síndrome do olho de gato, 1 (5,6%) com síndrome neuroectodérmica de Zunich, 1 (5,6%) com síndrome de CHARGE, e 1 (5,6%) com síndrome "morning glory" (Tabela 3).
Em 8 pacientes (44,4%) foi observado algum tipo de acometimento sistêmico. Entre as alterações sistêmicas foi encontrado acometimento do sistema nervoso central (SNC) em 4 (22,2%), retardo de crescimento em 2 (11,8%), hipoacusia em 2 (11,8%), fístula pré-auricular em 2 (11,8%), acometimento cardíaco em 1 (5,6%), fenda palatina em 1, acometimento cutâneo em 1, hérnia umbilical em 1, atresia de coanas em 1 e criptorquidismo em 1 único paciente.
A presença de coloboma também esteve associado a alterações oculares concomitantes. Em 14 pacientes (77,8%) foi encontrado algum tipo de alteração ocular além do coloboma. Em 8 pacientes (44,4%) estava associado estrabismo, em 6 (33,3%) nistagmo, 4 (22,2%) catarata, 1 paciente (5,6%) subluxação do cristalino e 1 paciente (5,6%) ceratocone.
DISCUSSÃO
No presente trabalho, foram estudados e avaliados 18 pacientes com coloboma ocular de um total de 1325 pacientes, representando 0,75% dos pacientes matriculados no serviço de Visão Subnormal do HSG. Em 1988 Kara José et al, em trabalho realizado em Campinas - SP, encontrou uma incidência de 4,3% de pacientes com coloboma atendidos no serviço de Visão Subnormal(4). Leal et al em 1995, em um estudo realizado em Recife - PE, verificaram uma incidência de 0,63% de colobomas(5). No nosso mesmo Serviço em 1997, Reis et al, encontram 0,5% de casos de coloboma ocular nos primeiros 435 pacientes(6). Destes 18 pacientes, 10 (55,6%) foram do sexo feminino. A literatura mostra uma prevalência de coloboma entre adultos cegos de 0,6-1,9%,(26) e entre crianças cegas a prevalência aumenta consideravelmente para 3,2-11,2%(7-8).
Quanto à raça, a leucodérmica foi a mais acometida com 15 casos (83,3%). Não existe consenso na literatura a respeito de sexo e raça com maior prevalência de colobomas, apresentando variações de acordo com a população estudada(8). O coloboma ocular típico, localizado no quadrante nasal inferior, foi o mais freqüente, ocorrendo em 77,8% dos olhos estudados, sendo o atípico observado em apenas 22,2% dos casos, fato concordante com a literatura(1).
Apesar do coloboma de íris poder se apresentar de forma isolada, freqüentemente é possível observar associação com outros colobomas. Dos 18 pacientes, 10 (55,6%) pacientes apresentavam coloboma de íris sendo que neste grupo em 100% dos casos houve associação com coloboma em outras regiões (zônula e corpo ciliar, retina, nervo óptico )(9).
Embora os colobomas possam estar associados a várias síndromes oculares(10-11) como síndrome de Goldenhar, síndrome de Rubinstein-Taybi, síndrome de Aicardi, síndrome de Meckel, síndrome de CHARGE, e outras, além de poder estar associado a anormalidades cromossômicas como trisomias, síndrome de Turner, síndrome de Klinefelter, síndrome do olho de gato, entre outras(10,12), no presente trabalho, em 9 pacientes (50%) os colobomas apresentaram a etiologia idiopática como a mais comum. A síndrome de olho de gato ocorreu em 6 casos (33,2%), síndrome de Zunich em 1 caso (5,6%), síndrome de CHARGE em 1 caso (5,6%) e síndrome de "morning glory" em 1 caso (5,6%).
A história gestacional se mostrou negativa em todos os pacientes. Em nenhum paciente foi possível identificar na história um agente que pudesse ter contribuído para o aparecimento de coloboma e/ou microftalmia. Todos negaram uso de drogas como talidomida, anticonvulsivantes (hidantoína, carbamazepina) durante a gestação, bem como negaram ocorrência de infecções embriopáticas como, citomegalovírus, Epstein-Barr, herpes e varicella-zoster. A exposição à irradiação, deficiência de ácido fólico e vitamina A, bem como ingestão de álcool durante a gestação foram negativas. Apesar de existirem inúmeros relatos de anomalias oculares associados às situações acima descritas como infecções virais(13-14) intoxicação alcoólica(15), uso de anticonvulsivantes, entre outras, a talidomida é a única substância comprovadamente causadora de coloboma em humanos(1,16).
A história familiar se mostrou positiva em apenas 3 casos, onde os três pacientes eram irmãos e tinham como diagnóstico a síndrome do olho de gato. Apesar disso, a história gestacional foi negativa com a mãe tendo passado a gravidez sem nenhuma intercorrência e sem ter usado qualquer medicação. Embora a síndrome seja caracterizada por anormalidade cromossômica, geralmente um cromossoma extra, os pacientes ainda estão em investigação, assim como os familiares para melhor esclarecimento dos casos(17).
No estudo foi encontrado microftalmia em 6 pacientes e nenhum caso de anoftalmia. A microftalmia é uma malformação congênita na qual se observa uma redução do volume ocular sempre em relação à faixa etária em estudo(18). Existe uma relação entre microftalmia e coloboma, sendo o coloboma freqüentemente associado a microftalmia, existindo uma relação embriológica na qual as causas de coloboma podem também ocasionar microftalmia(19).
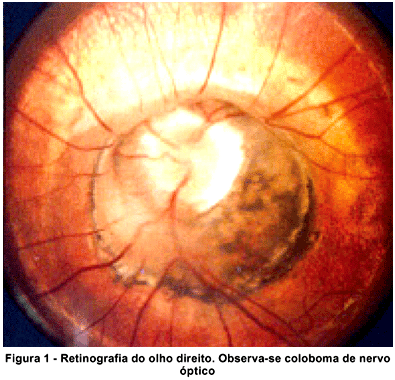
Em relação à estrutura ocular envolvida o coloboma retinocoroidal foi o mais comum ocorrendo em 16 pacientes ( 88,9%) estudados, seguido do coloboma de nervo óptico (Figura 1) com 14 (77,8%), coloboma de íris com 10 (55,6%), coloboma de mácula com 2 (11,1%) e coloboma de corpo ciliar e zônula com 1 (5,6%). Somente um paciente apresentou acometimento de apenas uma estrutura ocular, com coloboma de nervo óptico, todos os outros apresentaram mais de uma estrutura ocular envolvida. A incidência maior de colobomas do segmento posterior no presente trabalho está relacionada à população estudada, pacientes com visão subnormal, pois os colobomas do segmento posterior se relacionam aos piores prognósticos visuais(1). Um dos casos de coloboma de nervo óptico é a síndrome de "morning glory" considerada por alguns autores como um tipo de coloboma de nervo óptico atípico(1,20), sendo por outros autores considerada uma doença diferente e não um coloboma(21-22). Nesta anomalia de disco o mesmo se apresenta com aspecto grande, largo, com um proeminente anel peripapilar pigmentado e vasos emergindo de forma radial dando a aparência de uma flor de nome "morning glory".
Entre as complicações oculares relacionadas ao coloboma, no presente estudo foi possível observar que o estrabismo foi a complicação mais comum ocorrendo em 8 pacientes (44,4%), seguido do nistagmo com 6 (33,3%) casos, catarata com 4 casos, ceratocone com um caso e subluxação do cristalino também com um caso. Apesar do descolamento de retina ser uma das complicações mais comuns, relacionada principalmente ao coloboma retinocoroidal, sendo relatado na literatura uma incidência de 4-40% dos casos(23-24), não foi verificado neste levantamento retrospectivo. Não foi encontrado na literatura trabalhos com valores referentes à incidência de estrabismo, ambliopia e catarata em pacientes com coloboma, mas apenas relatos de casos de pacientes com coloboma e as referidas complicações(25-26). Mais uma vez estes achados estão relacionados à baixa acuidade visual apresentada por estes pacientes devido à alta incidência de colobomas acometendo o segmento posterior, principalmente retina e nervo óptico.
Foram observadas alterações sistêmicas em 8 pacientes (44,4%). Destes, 3 (16,7%) apresentavam síndrome do olho de gato, 1 ( 5,6%) paciente com síndrome de CHARGE, 1 ( 5,6%) com a síndrome neuroectodérmica de Zunich e nos outros 3 (16,7%) pacientes não foi possível estabelecer nenhum diagnóstico sindrômico.
O paciente com síndrome de CHARGE apresentava atresia de coanas, hipoacusia, defeito cardíaco, implantação baixa das orelhas e retardo de crescimento. A síndrome de CHARGE deriva-se do seu próprio nome. Para se estabelecer o diagnostico de CHARGE é preciso que o paciente apresente pelo menos 4 alterações determinadas pelas letras mnemônicas: coloboma, defeito cardíaco (Heart defect), atresia de coana (atresia of the choanae), retardo de crescimento, hipoplasia genital (Genital hypoplasia), e anomalias na orelha e/ou audição (Ear anomalies)(27). A maioria dos casos de CHARGE são esporádicos assim como o caso aqui presente. Acredita-se que a síndrome ocorra após um insulto durante o segundo mês gestacional(28-29).
O paciente com síndrome neuroectodérmica de Zunich apresentava além do coloboma retinocoroideano, acometimento no SNC com manifestações de epilepsia e retardo mental. Associado ao acometimento no SNC o paciente ainda apresentava acometimento cutâneo com ictiose vulgar e hiperceratose palmar. A síndrome neuroectodérmica de Zunich é uma das várias desordens que se enquadram no grupo das dermatoses ictiosiformes, e se caracterizam por apresentar acometimento neuroectodermico com erupção ictiosiforme, hipoacusia, retardo mental e epilepsia como as principais manifestações(30-32).
Nos pacientes com síndrome de olho de gato em dois casos foi encontrado acometimento no SNC sendo que em 1 caso foi observado ausência de IV ventrículo e no outro hipotrofia cerebral. Além disto foi encontrado fenda palatina em um caso; criptorquidismo e fístula pré-auricular em outro. Os pacientes com síndrome do olho de gato geralmente apresentam desenvolvimento mental normal ou próximo ao normal(33). Podem ser observadas anormalidades cromossômicas ou não. Microftalmia, estrabismo, ânus imperfurado, fístula pré-auricular, anomalias renais e cardíacas podem ocorrer(17).
A acuidade visual variou nos pacientes desde ausência de percepção luminosa até 0,4 (20/50) com média de 0,1 (20/200). Pode-se notar, portanto, que os valores de acuidade visual variaram de uma visão próxima ao normal até cegueira total, com uma média de pacientes apresentando baixa visão severa. A prescrição de auxílios ópticos especiais depende de fatores como acuidade visual, idade do paciente, necessidades definidas, bem como, a capacidade de resposta ao auxílio, fatores estes, que justificaram o número de auxílios prescritos.
Os colobomas podem apresentar um acometimento visual bastante variado, desde casos com visão normal até a cegueira total dependendo da extensão e localização dos mesmos. É fundamental que as crianças com coloboma e suspeita de baixa visão sejam avaliadas por oftalmologistas com especialização em baixa visão para que possa ser feito o diagnóstico e orientações apropriados para que, então, o paciente possa receber toda informação, treinamento e prescrição de magnificação que se fizer necessário.
CONCLUSÃO
A avaliação de pacientes com coloboma deve ser realizada muito cuidadosamente. Todas os pacientes devem ser meticulosamente examinados, mesmo crianças que são pouco cooperativas, com intuito de se procurar diferentes localizações para os colobomas. O exame refracional é essencial e de grande valor visto o risco de ambliopia e anisometropia que pode ocorrer nestes pacientes. Oftalmoscopia direta e indireta deve ser realizada sempre para descartar a presença de colobomas coriorretinianos e/ou de nervo óptico. Exames complementares como tomografia de crânio devem ser realizados quando existe suspeita de acometimento no SNC.
O coloboma ocular assim como a microftalmia são malformações congênitas comuns que podem estar associadas ou não. Na maior parte dos casos se apresenta como uma alteração ocular isolada, podendo em algumas situações estar associada a outras anomalias ou síndromes. Colobomas associados a outras anormalidades sistêmicas devem ser submetidos a estudo cromossômico da família para complementação diagnóstica, prognóstica e para aconselhamento genético.
AGRADECIMENTO
A Dra. Márcia Guimarães pelo apoio científico e pela foto cedida para este trabalho.
REFERÊNCIAS
1. Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol 2000;45:175-94.
2. Duke-Elder S. System of ophthalmology. St. Louis: Mosby; 1963. v.3, p. 456-709.
3. Barber AN. Embryology of the human eye. St. Louis: Mosby; 1995. p.50-63.
4. Kara José N, Carvalho KMM Pereira VL, Venturini NHB, Gasparetto MEFR, Gushiken MT. Estudo retrospectivo dos primeiros 140 casos atendidos na clínica de visão sub-normal do Hospital de Clínicas da UNICAMP. Arq. Bras. Oftalmol 1988;51:65-9.
5. Leal DB, Tavares SS, Ventura LO, Florêncio T. Atendimento a portadores de visão subnormal: estudo retrospectivo de 317 casos. Arq Bras. Oftalmol 1995;58: 439-42.
6. Reis PAC, Campos CMC, Fernandes LC. Características da população portadora de visão subnormal do Hospital São Geraldo: um estudo retrospectivo de 435 casos. Rev. Bras. Oftalmol 1998;57:287-94.
7. Fraser Gr, Friedman AL: The causes of blindness in childhood. Baltimore: John Hopkins Press; 1967.
8. Fujika K, Makajima A, Yasuda N, Tanaka U, Kabasawa K. Genetic analysis of microphthalmos. Ophthalmic Paediatr Genet 1982;1:139-49.
9. Cagianut B, Theiler k: Bilateral colobomas of iris and choroid. Association with partial deletion of a chromosome of group D. Arch Ophthalmol 1970;83:141-4.
10. Basic and clinical science course 2000-2001: Pediatric ophthalmology and strabismus. American Academy of Ophthalmology; 2000. Section 6, p.219–20.
11. Hornby SJ, Gothwal VK, Adolpg S, Gilbert CE. The needs of children with congenital eye anomalies (Microphthalmos and Coloboma) for low vision services in India. In: Stuen C, Arditi A, Horowitz A et al. Vision rehabilitation assessment, intervention and outcomes. Nova York: Swets & Zeitlinger; 2000. p.362-6.
12. Wilson ME, O'Neil JW. Pediatric iris abnormalities. In: Wright KW, ed. Pediatric Ophthalmology and Strabismus. St Louis; 1995. p.349-65.
13. Frenkel LD, Keys MP, Hefferen SJ, Rola-Pleszczynski M, Bellanti JA. Unusual eye abnormalities associated with congenital cytomegalovirus infection. Pediatrics 1980;66:763-6.
14. Goldberg GN, Fulginiti VA, Ray CG, Ferry P, Jones JF, Cross H, Minnich L. In utero Epstein-Barr virus (infectious mononucleosis) infection. JAMA 1981; 246:1579-81.
15. Strömland K. Ocular involvement in the fetal alcohol syndrome. Surv Ophthalmol 1987;31:277-84.
16. Smithells RW. Defects and disabilities of thalidomide children. Br Med J 1973;1:269-72.
17. Walknowska J, Peakman D, Weleber RG. Cytogenetic investigation of cat eye syndrome. Am J Ophthamol 1977;84:477-86.
18. Bateman JB: Microphthalmos. Int Ophthalmol Clin 1984;24:87-107.
19. Elder MJ: Aetiology of severe visual impairment and blindness in microphthalmos. Br J Ophthalmol 1994;78:332-4.
20. Eustis HS, Sanders MR, Zimmerman T: Morning glory syndrome in children. Association with endocrine and central nervous system anomalies. Arch Ophthalmol 1994;112:204-7.
21. Apple DJ, Rabb MF, Walsh PM. Congenital anomalies of the optic disc. Surv Ophthalmol 1982;27:3-41.
22. Brodsky MC, Baker RS, Hamed LM. Pediatric neuro-ophthalmology. New York: Springer;1996. p.63.
23. Irvine AR, Crawford JB, Sullivan JH. The pathogenesis of retinal detachment with morning glory disc and optic pit. Retina 1986;6:146-50.
24. Brown GC, Brown MM. Repair of retinal detachment asociated with congenital excavated defects of the optic disc. Ophthalmic Surg 1995; 26:11-5.
25. Antle Cm, Pantzar JT, White VA. The ocular pathology of trisomy 22: report of two cases and review. J Pediatr Ophthalmol Strabismus 1990;27:310-4.
26. Fulton AB, Howard RO, Albert DM, Hsia YE, Packman S. Ocular findings in triploidy. Am J Ophthalmol 1977;84:859-67.
27. Chestler RJ, France TD. Ocular findings in CHARGE syndrome. Six case reports and a review. Ophthalmology 1988;95:1613-9.
28. Russell-Eggitt IM, Blake KD, Taylor DS, Wyse RK. The eye in the CHARGE association. Br J Ophthalmol 1990,74:421-6.
29. Warburg M. Ocular coloboma and multiple congenital anomalies: the CHARGE association. Ophthalmic Pediatric Genet 1983,2:189-93
30. Baden HP, Bronstein BR. Ichthyosiform dermatosis and deafness. Report of a case and review of the literature. Arch Dermatol 1988;124:102-6.
31. Zunich J, Esterly NB, Holbrook KA, Kaye CI. Congenital migratory ichthyosiform dermatosis with neurologic and ophthalmologic abnormalities. Arch Dermatol 1985;121:1149-56.
32. Zunich J, Kaye CI. New syndrome of congenital ichthyosis with neurologic abnormalities. Am J Med Genet 1983;15:331-3, 5.
33. Cory CC, Jamison DL. The cat eye syndrome. Arch Ophthalmol 1974; 92:259-62.
Endereço para correspondência
Patrick Frensel de Moraes Tzelikis
Rua Pernambuco, nº 439/1002
Belo Horizonte (MG) - CEP 30130-150
E-mail: [email protected]
Recebido para publicação em 29.10.2002
Versão revisada recebida em 06.03.2003
Aprovação em 10.03.2003
Trabalho realizado no Hospital São Geraldo da Universidade Federal de Minas Gerais - UFMG.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. Marcos Pereira de Ávila.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.