Arq. Bras. Oftalmol. 2003; 66 (4): 10.1590/S0004-27492003000400002
Total: 2625
Célia Beatriz Gianotti Antoneli1; Flávio Steinhorst2; Karina de Cassia Braga Ribeiro3; Clélia Maria Erwenne4; Paulo Eduardo R.S. Novaes5; Victor Arias6; Alois Bianchi2
DOI: 10.1590/S0004-27492003000400002
RESUMO
OBJETIVO: Mostrar a evolução do tratamento do retinoblastoma ao longo de uma década e suas implicações na sobrevida global e na preservação da visão. MÉTODOS: Trezentos e dezenove pacientes foram avaliados no estudo. Analisamos 257 pacientes portadores de retinoblastoma admitidos no Hospital do Câncer, entre janeiro de 1986 a dezembro de 1996, divididos em dois grupos de acordo com tratamento a que foram submetidos (1986 a 1990 e 1991 a 1996). Analisamos ainda 62 pacientes portadores de retinoblastoma intra-ocular entre janeiro de 1996 e maio de 2000 que foram submetidos a quimiorredução, aliada a medidas oftalmológicas, sem enucleação ou radioterapia externa. RESULTADOS: Em pacientes com doença intra-ocular, nos dois períodos, não houve significância estatística com relação à sobrevida. Analisamos 59 pacientes com retinoblastoma extra-ocular, de acordo com a classificação do CCG (Children´s Cancer Group), e a sobrevida global em 5 anos foi maior no 2º período, (63,7% vs. 41%; p= 0,059). Entre 1996 e 2000 analisamos 62 pacientes com retinoblastoma intra-ocular com visão preservada, sendo 47 (75,8%) com doença bilateral e 15 (24,2%) com tumor unilateral. Os portadores de tumor bilateral tiveram uma taxa de preservação de visão de 49,8%, versus 26,7% para os pacientes com tumor unilateral. CONCLUSÕES: Tumores intra-oculares mantêm altas taxas de cura e preservação da visão. Novas combinações de drogas poderão melhorar a sobrevida dos extra-oculares. Quanto às perspectivas futuras, há pesquisas envolvendo terapia gênica/molecular e terapêutica anti-angiogênese. Apesar de toda a evolução, o diagnóstico precoce é a arma mais poderosa, na cura dos portadores deste tumor.
Descritores: Retinoblastoma; Sobrevivência; Visão
ABSTRACT
PURPOSE: The management of retinoblastoma (Rb) has gradually changed over the past years achieving a satisfactory result (increasing overall survival and avoiding enucleations). METHODS: Three hundred and nineteen patients with retinoblastoma were studied. From January 1986 to November 1996, 257 patients with Rb were referred to "Hospital do Cancer AC Camargo". The treatment changed according to the period, from 1986 to 1990 and from 1991 to 1996. From January 1996 to December 2000, 62 patients presenting intraocular (IO) tumors were treated with chemoreduction and adjuvant treatment to avoid enucleation or external beam radiation therapy. RESULTS: From January 1996 to December 2000, 62 patients with IO Rb (47 bilateral and 15 unilateral tumors) with potential vision were treated with chemoreduction and ophthalmological adjuvant treatment. In the patients with bilateral tumors 49.8% preserved the eye vs. 26.7% for patients with unilateral tumors. Among 257 patients 59 presented extraocular Rb according to CCG classification. Overall five-year survival was improved in the second period (63.7 x 41% - p=0.059). CONCLUSION: Chemorreduction plus focal treatment plays an important role in the management of children with Rb. Gene therapy perhaps will increase the number of preserved eyes. A multicenter trial should be considered to evaluate patients with CNS Rb or metastatic disease with ominous prognosis. The lag time between the first sign and symptoms and the early diagnosis is the best weapon to achieve the cure in this patients.
Keywords: Retinoblastoma; Survival; Vision
INTRODUÇÃO
O sucesso na abordagem terapêutica dos pacientes com retinoblastoma requer um enfoque multidisciplinar(1). Atenção especial dos pediatras na puericultura, quanto a alterações oculares (estrabismo, leucocoria - mancha branca na íris conhecida como "olho de gato") que podem alertar para o diagnóstico precoce. O reconhecimento destas alterações e o encaminhamento para o oftalmologista são fundamentais para o sucesso do tratamento desta doença.
O retinoblastoma é o tumor maligno intra-ocular mais freqüente na criança, correspondendo a 2 a 4% dos tumores malignos pediátricos. Tem uma incidência de 11 casos novos por milhão em menores de 5 anos nos Estados Unidos (1:18.000 nascidos vivos = 200 casos/ano), sem diferença entre sexo ou raça, é o 4º tumor mais freqüente em crianças até 14 anos de idade (Brasil), 80% são diagnosticados antes dos 3-4 anos de idade e 30 a 40% são bilaterais(2).
Os sintomas que podem chamar a atenção para o diagnóstico precoce do retinoblastoma levando ao encaminhamento ao oftalmologista são: reflexo do "olho de gato" (leucocoria - Fig. 1), massa orbitária, estrabismo, cor diferente dos olhos (heterocromia), hiperemia conjuntival, cefaléia, vômitos, dor óssea e perda de visão.
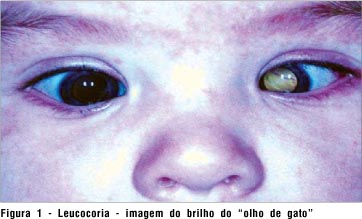
O diagnóstico definitivo pode ser feito através de um exame de fundo de olho (sob anestesia geral), e os demais exames com a finalidade do estadiamento para classificar o tumor e direcionar o tratamento - tomografia de crânio e órbita, ultra-sonografia do olho, raio-x de crânio, mielograma e citológico do líquido cefalorraquidiano.
Com relação aos pacientes portadores de tumores intra-oculares, até o final da década de 70, a maioria dos autores preconizava a enucleação e, eventualmente, radioterápico. Com esses métodos terapêuticos, os pacientes apresentavam uma sobrevida de 90%(3).
A partir dos anos 80, o uso de drogas quimioterápicas adjuvantes a enucleação, limitava-se àqueles pacientes com reconhecidos fatores de risco para doença micrometastática como: volume do tumor (> ou igual a 10% do volume do bulbo ocular), comprimento do coto do nervo óptico (< ou igual a 5 mm), extensão da invasão coroidal e grau de diferenciação celular(4).
Na década de 90, foi proposto um protocolo de quimioredução com Carboplatina, Etoposide e Vincristina, para portadores de retinoblastoma intra-ocular com visão, com o objetivo de preservar o bulbo ocular e a própria acuidade visual(5).
O tratamento oftalmológico local, leva em conta o tamanho e a localização das lesões, em se tratando de tumores intra-oculares. Modalidades de tratamento local como placa radioativa, crioterapia, termoterapia, e fotocoagulação com laser, têm sido usadas com sucesso para tratar tumores intra-oculares menores, mas são menos efetivos em lesões mais avançadas(6-8).
Na tentativa de evitar a enucleação e radioterapia externa, com suas morbidades associadas, preconiza-se a quimiorredução, para redução do volume tumoral e otimização da terapêutica oftalmológica local(9).
Com relação aos tumores extra-oculares (que se estendem além do bulbo ocular), poucos relatos são encontrados na literatura, embora se saiba que nos países em desenvolvimento esse tipo de doença é mais freqüente do que nos países desenvolvidos(9).
A quimioterapia tem se mostrado efetiva no tratamento de doença extra-ocular, quando esses tumores não são metastáticos(10-11).
O seguimento pós-tratamento visa identificar recorrências. Neste tipo de tumor ela ocorre com maior freqüência nos 3 primeiros anos após diagnóstico. Recaída após 5 anos é rara. Além de recorrência o seguimento dos pacientes é importante para o diagnóstico de efeitos tardios e um possível segundo tumor(12). O prognóstico destes tumores é bastante variado, com íntima relação ao estádio da doença(3).
OBJETIVO
O objetivo deste estudo é mostrar a evolução do tratamento quimioterápico e oftalmológico do retinoblastoma ao longo de uma década e suas implicações na sobrevida global e preservação da visão, e ainda, demonstrar, através de uma breve revisão, os avanços e novas perspectivas no tratamento de crianças com retinoblastoma.
MÉTODOS
Trezentos e dezenove pacientes fizeram parte do estudo.
Analisamos 257 pacientes portadores de retinoblastoma admitidos nos departamentos de Pediatria e Oftalmologia do Hospital do Câncer, no período de janeiro de 1986 a dezembro de 1995. Analisamos ainda, 62 pacientes portadores de retinoblastoma intra-ocular admitidos no período compreendido entre janeiro de 1996 a maio de 2000.
Para os pacientes portadores de tumores intra-oculares adotamos o esquema de estadiamento preconizado por Reese-Ellseworth(13), (Tabela 1) e para os portadores de doença extra-ocular, adotamos a classificação do CCG (Tabela 2)(14).
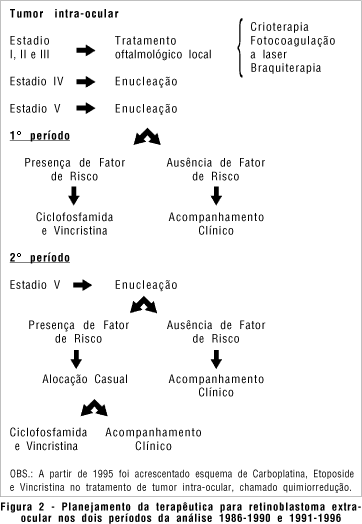
Ao longo desses anos, os pacientes foram divididos em dois períodos de acordo com a proposta terapêutica: primeiro período compreendido entre janeiro de1986 dezembro de 1990 e segundo período compreendido entre os anos de 1991 a 1995. No primeiro período da análise, todos os pacientes portadores de tumor intra-ocular Reese V eram submetidos a enucleação e, se apresentassem pelo menos um fator de risco (tamanho do tumor > a 10% do volume do globo ocular, tumor além da lâmina cribosa, ou coto do nervo óptico < a 5 mm), eram submetidos a tratamento quimioterápico com Ciclofosfamida (30 mg/kg/dia Dia 1 (D1) e Vincristina (0,05 mg/kg/dia D1) a cada 21 dias, perfazendo um total de 10 ciclos de tratamento).
No segundo período da análise, os pacientes portadores de retinoblastoma classificados como Reese-Ellsworth V, com fator de risco presente, foram sorteados para receber, ou não, tratamento quimioterápico.
No intuito de avaliar os casos que não foram enucleados e nem receberam radioterapia externa, analisamos 62 pacientes com retinoblastoma intra-ocular e visão conservada, no período compreendido entre janeiro de 1996 a maio de 2000. Os mesmos foram submetidos a tratamento quimioterápico com Carboplatina 200 mg/m2/ dia do dia 1 ao dia 3 ( D1-3), Etoposide 150 mg/m2/dia D1-3 e Vincristina 1,5 mg/m2/dia D1 a cada 21 dias até um total de 6 ciclos, associado a medidas oftalmológicas como crioterapia, fotocoagulação com laser e braquiterapia quando indicada. Os casos refratários seguiam tratamento adicional com radioterapia externa e/ou enucleação.
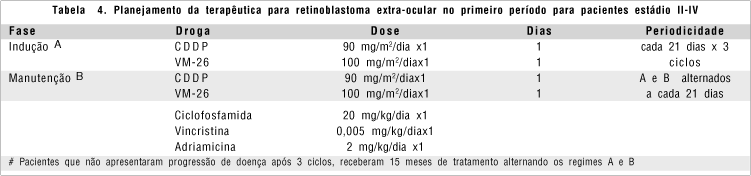
Cinqüenta e nove pacientes portadores de tumor extra-ocular foram analisados separadamente, visto que a abordagem também sofreu mudanças ao longo dessa década de estudo, sendo analisados de acordo com o período em que foram admitidos na Instituição. O esquema terapêutico preconizado no primeiro período da análise (janeiro de 1986 a dezembro de 1990), utilizava as seguintes drogas: Cisplatina e Teniposide alternadas com Ciclofosfamida, Vincristina e Doxorubicina. No segundo período, entre os anos de 1991 a 1995, adicionou-se ao esquema acima uma janela terapêutica com Ifosfamida 1800mg/m2/dia do dia 1 ao dia 5 (D1-5) e Etoposide 100mg/m2/dia D1-5, por três ciclos e, posteriormente prosseguia-se com o mesmo esquema terapêutico do primeiro período da análise, alternando Ifosfamida e Etoposide, perfazendo um total de 15 meses de tratamento (ver planejamento da terapêutica nas figuras 2 e 3).

Criamos um banco de dados para registro e arquivo das informações coletadas para posterior processamentos estatísticos, utilizando-se o programa D Base III Plus.
A análise estatística comporta a estatística descritiva e teste do Qui-quadrado. Para evitar distorções na apreciação da significância estatística o teste do Qui-quadrado foi substituído pelo teste exato de Fisher, sempre que necessário. A análise da sobrevida global foi calculada pelo método de Kaplan Méier.
Comparações entre as distribuições de sobrevida para uma mesma variável foram feitas pelo teste comparativo de Cox(15).
Definimos sobrevida global como o tempo em meses, decorrido entre o início do tratamento e o óbito.
RESULTADOS
Analisamos 257 pacientes portadores de retinoblastoma, 198 (77%) eram portadores de tumores intra-oculares, 3 foram classificados como RE II, 2 com RE III, 6 como RE IV e 187, como RE V. Para efeito de tratamento, somente os pacientes portadores de tumores estadio V intra-ocular e os extra-oculares, farão parte da análise. No período entre 1986 e 1990, 71/78 pacientes foram submetidos a tratamento quimioterápico e no período compreendido entre 1991 a 1995, 58/120 receberam a combinação de drogas preconizada. Os resultados obtidos no segundo período da análise não mostraram significância estatística entre os pacientes que receberam e os que não receberam quimioterapia com relação à sobrevida. Esse fato nos leva a pensar que talvez os fatores descritos acima, não sejam considerados como de risco para doença micrometastática e a quimioterapia adjuvante não seja necessária para os pacientes que preencham esses critérios(15).
Independente do período, na casuística do Hospital do Câncer, com 257 pacientes estudados, demonstramos uma sobrevida > 90% para tumor intra-ocular, 77% para tumor extra-ocular I a III e, para pacientes com doença no líquor ou medula óssea (IV e V) a sobrevida média é de 13 meses, sem nenhum paciente vivo após esse período de tempo.
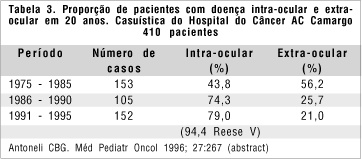
A proporção de pacientes com doença intra-ocular e extra-ocular foi a mesma nos dois períodos da análise (1986 a 1990 e 1991 a 1995). Para pacientes com doença intra-ocular, a porcentagem de casos no primeiro período foi de 74,3% vs. 79% no segundo. Para pacientes com doença extra-ocular, a porcentagem foi de 25,7% no primeiro período vs 21%, no segundo período da análise. Quando comparamos estes dados com os de 158 pacientes analisados anteriormente, no período de 1975 a 1985(16), onde 56,2% eram portadores de doença intra-ocular e 43,8%, de retinoblastoma extra-ocular, pudemos observar que houve uma redução no número de pacientes com doença avançada nos últimos vinte anos (Tabela. 3).
Também analisamos, num terceiro período (1996 a 2000), os pacientes com doença intra-ocular com visão preservada que foram submetidos à intensificação de quimioterapia aliada a medidas oftalmológicas e placa radioativa, na tentativa de evitar a enucleação e a radioterapia externa. Sessenta e dois pacientes foram avaliados, sendo que 47 (75,8%) apresentavam doença bilateral e 15 (24,2%), tumor unilateral. Com o emprego da quimiorredução mais terapêutica oftalmológica local (laserterapia, fotocoagulação) mais placa de Cobalto na dependência do tamanho e localização da lesão, 49,8% dos pacientes com tumores bilaterais e 26,7% dos pacientes com doença unilateral, tiveram preservação da visão sem que se fizesse necessária radioterapia externa(17).
Quanto aos casos de retinoblastoma avançados, 59 (23%) pacientes portadores de doença extra-ocular, foram avaliados, no período compreendido entre janeiro de 1986 a dezembro de 1996. A sobrevida global em 5 anos foi maior no 2º período, compreendido entre 1991 e 1996, comparada ao período anterior (63,7% vs. 41%; p= 0,059). Esse fato foi atribuído ao uso da combinação Ifosfamida e Etoposide adicionada ao esquema terapêutico preconizado anteriormente1 (Acreditamos que com um "n" maior o "p" seja significativo) ou aos avanços nas medidas de suporte e manejo adequado das complicações do tratamento(18).
Os pacientes com doença extra-ocular, avançada localmente, (vasos emissários esclerais comprometidos, tumor ao nível da secção do nervo óptico ou presença de tumor em órbita), ou seja, tumores classificados de acordo com o CCG como classe I, II e III, tiveram uma melhor sobrevida global em 5 anos no período considerado a partir de 1991 (77,5% vs.53,6%; p = 0,05). Quando consideramos doença bilateral, observa-se que os portadores de tumor localmente avançado apresentaram melhor sobrevida que os pacientes com tumor unilateral (92,9% vs. 49,6%; p= 0,01). Infelizmente os pacientes com doença em sistema nervoso central, metástase óssea ou em medula óssea mantiveram um prognóstico sombrio, evoluindo para um êxito letal, com um tempo médio de sobrevida de 13 meses(18).
DISCUSSÃO
Com avanços na terapêutica, a sobrevida dos pacientes portadores de retinoblastoma tem passado de 30%, em 1930 para 95% em 1990(19). Até poucos anos atrás, enucleação e radioterapia externa eram as modalidades de escolha no tratamento do retinoblastoma e foram responsáveis por esta marcada melhora da sobrevida.
Um fato a ser mencionado é que essas modalidades terapêuticas são também associadas com significante morbidade, especialmente seqüelas crânio-faciais, em grande parte pela enucleação e ação da radioterapia externa, que pode, ainda ser responsabilizada pelo aparecimento de segundo tumor em área irradiada(19-20). Com os novos conhecimentos a respeito da doença e com a descoberta de novas modalidades terapêuticas, cada vez mais visam diminuir a morbidade e aumentar, ainda mais, as taxas de sobrevida.
A partir dos anos 90, passou-se a divulgar o uso de tratamento quimioterápico para pacientes portadores de retinoblastoma intra-ocular com finalidade de se evitar o tratamento cirúrgico.
Alguns autores demonstraram sucesso terapêutico em paciente portador de retinoblastoma recorrente, com implante de células vítreas, utilizando a combinação de Ranimustina (MCNU) e Carboplatina(21), enquanto outros, utilizaram-se da Carboplatina mais terapia local (fotocoagulação e crioterapia) como modalidades de tratamento alternativo à radioterapia externa, em pacientes com tumores intra-oculares, demonstrando que a radioterapia externa pode ser evitada como tratamento primário em pacientes com retinoblastoma Reese-Ellsworth grupos I a IV (22).
Estudo piloto prospectivo com 20 pacientes com tumor intra-ocular, preconizando quimioredução com Carboplatina, Etoposide e Vincristina, durante dois meses a fim de avaliar a redução do tumor, possibilitando o emprego de tratamento conservador com medidas oftalmológicas associadas, tais como termoterapia foi também proposto. A conclusão desse estudo foi que houve redução no tamanho do tumor e que medidas menos invasivas como crioterapia, fotocoagulação, termoterapia e placa radioativa puderam ser utilizadas, evitando tanto a enucleação quanto à radioterapia externa(5). Outros autores corroboram estes dados(23-25).
Conforme apontamos nos nossos resultados, o uso de quimioredução vem se mostrando promissor. Conseguimos preservar a visão em quase 50% dos pacientes com retinoblastoma bilateral e em ¼ dos pacientes com tumor unilateral. A toxicidade ao esquema quimioterápico foi aceitável.
A análise de 814 prontuários de pacientes portadores de retinoblastoma, no período de 1922 a 1986, com pelo menos um dos olhos enucleados, descreveu 240 (29,5%) pacientes com envolvimento do nervo óptico e concluiu que os pacientes que apresentavam comprometimento além da lâmina cribosa tinham uma taxa de mortalidade maior quando comparados aos pacientes que não apresentavam este achado anátomo-patológico(26).
Até então, o que é descrito como de alto risco para metastatização são: comprometimento de segmento anterior, infiltração de íris, esclera ou corpo ciliar, infiltração maciça de coróide, invasão de nervo óptico (lâmina cribosa, retrolaminar ou ao nível da transecção) e extensão extra-escleral(27).
Quanto aos tumores extra-oculares, a literatura é pobre. Em países em desenvolvimento, a sobrevida global dos portadores de retinoblastoma extra-ocular gira ao redor de 44%(28). O mesmo autor, analisando 94 pacientes negros sul africanos portadores de retinoblastoma, entre os anos de 1989 a 1996, observou que em 84% destes pacientes a doença não era passível de tratamento conservador e que 34% apresentavam doença metastática ao diagnóstico(28).
O grande desafio é saber o que desencadeia o aparecimento de tumores avançados. Bolsões de miséria, nível nutricional pior, maior taxa de infecção, fatores ligados a maior exposição da retina à luz solar? O baixo nível cultural, a desinformação, o longo período tempo decorrido entre o primeiro sinal e sintoma até a chegada a um centro especializado? Os últimos são os principais fatores para o aparecimento de tumores extra-oculares.
A abordagem terapêutica ideal para crianças com retinoblastoma extra-ocular ainda apresenta muitos desafios e enigmas. São necessários critérios para identificar fatores de risco para recidiva e da necessidade ou não do emprego de quimioterapia sistêmica direcionada para cada um dos riscos. Pacientes com doença avançada que evoluíam rapidamente para óbito no passado têm experimentado uma melhora na sobrevida livre de eventos com a instituição de múltiplas modalidades de terapia de consolidação com transplante autólogo de medula óssea(29).
Embora tenhamos melhorado muito a sobrevida dos pacientes com tumores localmente avançados (estádios I, II e III extra-ocular), de 40,8% para 63,7%, temos um longo caminho a percorrer quando nos deparamos com tumores metastáticos ao diagnóstico.
A mais efetiva e menos tóxica combinação de agentes quimioterápicos e a duração de tratamento requer mais estudos. Pacientes com doença metastática e com doença extracoróide (alto risco para recaída) foram tratados com 3 ciclos de Carboplatina/Etoposide alternados com 3 ciclos de Ciclosporina/Doxorubicina/Carboplatina com resgate de G-CSF (fator estimulante de granulócitos) pós quimioterapia (10 a 14 dias). Após dois ciclos de quimioterapia os pacientes receberam radioterapia com mega eletron volt (MEV) em sítios de metástase. Pacientes com doença em medula óssea ao diagnóstico receberam transplante autólogo no final dos seis ciclos de quimioterapia. Esta abordagem foi eficaz quanto à resposta completa nos casos de doença metastática e na prevenção desta, em pacientes com doença extracoróide(30).
Não há consenso na literatura quanto ao melhor esquema quimioterápico para o tratamento do retinoblastoma extra-ocular. A literatura nos mostra autores que(31) analisaram 33 pacientes com envolvimento orbitário durante o período de 1977 a 1991, tratados com Ciclofosfamida, derivados da Platina, Epipodofilotoxinas, Doxorubicina e Vincristina concluindo que esta combinação foi eficaz. Um protocolo terapêutico com Carboplatina, Etoposide e Ciclofosfamida (CARBOPEL), seguido de resgate de células tronco autólogas, para pacientes com retinoblastoma extra-ocular ao diagnóstico ou recorrente, ou com invasão de coto de nervo óptico foi proposto por autores franceses. Com esse protocolo os mesmos observaram em 25 pacientes uma sobrevida livre de doença, em 3 anos, de 67,1%(32).
Os múltiplos esquemas para o tratamento do retinoblastoma têm estado ligados à grande morbidade. Segundo tumor ocorre naqueles pacientes com retinoblastoma hereditário com maior freqüência em relação a outras neoplasias pediátricas(6) e estão relacionados com tratamentos envolvendo quimioterapia e radioterapia, além da predisposição genética. Sarcomas ósseos e sarcomas de partes moles são os mais comuns após o retinoblastoma, seguido de melanoma, leucemia, linfoma e câncer de mama(33-34). A escolha da modalidade terapêutica deve, portanto, incluir a análise de fatores de risco genético para segunda neoplasia, e, a morbidade deverá ser devidamente considerada, bem como a vigilância destes pacientes por um longo período de tempo.
Baseados nisto, o principal enfoque na terapia anticâncer é erradicar as células tumorais sem comprometimento das células normais, o que vem se obtendo através de pesquisas envolvendo terapia gênica/molecular. Na verdade, com a descoberta da estrutura do DNA, a biologia molecular tem permitido o entendimento das diversas doenças genéticas e aberto perspectivas para a possibilidade de terapia gênica. A princípio, todas as células tumorais para sobreviver e proliferar adquirem mutações no gene supressor de tumor p53, que induz apoptose em resposta a algum estímulo oncogênico. Nos casos de retinoblastoma a proteína p53 selvagem é funcionalmente inibida. O p53 pode ser reativado seletivamente para induzir apoptose em células do retinoblastoma sem dano aos tecidos normais e com evidente resposta clínica e histopatológica da dissolução do tumor em 48 horas(35).
Para evitar morbidade associada ao tratamento de crianças com retinoblastoma, tem sido proposta a terapia molecular com genes suicidas, usando um vetor adenoviral (extraído do herpes simples). Esse processo demonstrou sucesso na regressão tumoral em estudos com ratos(36).
Barrios et al., (36) demonstraram num ensaio clínico fase I em 2 crianças com retinoblastoma que não há toxicidade limitante para esta modalidade de terapia gênica e na análise histopatológica pós enucleação não houve evidência de tumor viável.
A terapia gênica é ainda uma questão a ser discutida do ponto de vista médico, jurídico e filosófico, mas podemos vislumbrar seu amplo uso num futuro não muito distante. A desinformação cultural de pais e técnica por parte dos pediatras e oftalmologistas leva ao diagnóstico tardio e, em conseqüência, à presença de tumores avançados. O grande desafio está no diagnóstico precoce.
A partir de 1986, teve início uma campanha educativa para médicos e não-médicos, a qual acreditamos ter sido responsável pela diminuição da incidência de doença extra-ocular(15) (fig. 4).

Quando existe desconhecimento relacionado a algum determinado tipo de doença, a idade ao diagnóstico é mais avançada, retardando o tempo de encaminhamento entre o primeiro sinal e sintoma até um centro especializado. O diagnóstico deixando de ser precoce faz com que a doença se torne avançada e os índices de cura diminuam.
Quanto mais precoce o diagnóstico, menor a extensão da doença, maior as taxas de cura e há uma redução importante tanto nas seqüelas, quanto nos efeitos colaterais, decorrentes da terapêutica, seja a curto ou em longo prazo. O diagnóstico precoce é um dos principais aliados na determinação da cura do paciente portador de retinoblastoma.
CONCLUSÃO
Nos últimos 20-30 anos a abordagem do retinoblastoma vem mudando radicalmente. A princípio, os tumores intra-oculares eram tratados ou com enucleação ou radioterapia externa e não se permitia outra opção terapêutica. Com o emprego da quimioredução para tratamento desses tumores, hoje, um número considerável de pacientes, consegue manter a visão, sem os efeitos colaterais tardios decorrentes do emprego muitas vezes desnecessário, da radioterapia externa. A exemplo das leucemias e tumor de Wilms(37), o retinoblastoma é uma das neoplasias que tiveram melhora da sobrevida com o emprego de quimioterapia.
Para os portadores de tumores extra-oculares, novos esquemas quimioterápicos são necessários para o aumento da sobrevida, bem como, o avanço nas áreas da engenharia genética e terapia gênica.
Com todos os avanços da Medicina, o diagnóstico precoce do portador de retinoblastoma ainda é o fator fundamental para o bom êxito da terapêutica e da sobrevida.
REFERÊNCIAS
1. Antoneli CBG. Retinoblastoma: análise da evolução clínica de pacientes portadores de retinoblastoma submetidos a tratamento multidisciplinar . São Paulo: Faculdade de Medicina da Universidade de São Paulo; 1999.
2. Parkin DM, Stiller CA, Draper GJ, Bieber CA. The international incidence of childhood cancer. Int J Cancer 1988;42:511-20.
3. Donaldson SS, Egbert PR, Newsham I, Cavenee WK. Retinoblastoma. In: Pizzo PA, Poplack DG, editors. Principles and practice of Pediatric Oncology. 3rd ed. Philadelphia: Lipincott-Haven; 1997. p. 699-715.
4. White L. The role of chemotherapy in the treatment of retinoblastoma. Retina 1983;3:194-9.
5. Shields CL, Shields JA, De Potter P, Himelstein BP, Meadows AT, Maris JM. Chemoreduction in de initial management of intraocular retinoblastoma. Arch Ophthalmol 1996;114:1330-8.
6. Shields CL, Shields JA. Recent developments in the management of retinoblastoma. J Pediatr Ophthalmol Strabismus 1999;36:8-18.
7. Shields JA, Shields CL, De Potter P, Hernandez JC, Brady LW. Plaque radiotherapy for residual or recurrent retinoblastoma in 91 cases. J Pediatr Ophthalmol Strabismus 1994;31:242-5.
8. Shields JA, Shields CL, De Potter P, Minelli S, Hernandez C, Brady LW, Cater JR. Plaque radiotherapy in the management of retinoblastoma. Use as a primary and secondary treatment. Ophthalmology 1993;100:216-24.
9. Grabowski EF, Abramson DH. Intraocular and extraocular retinoblastoma. Hematol Oncol Clin North Am 1987;1:721-35.
10. Kingston JE, Hungerford JL, Madreperla SA, Plowman PN. Results of combined chemotherapy and radiotherapy for advanced intraocular retinoblastoma. Arch Ophthalmol 1996;114:1339-43.
11. Friedman DL, Himelstein B, Shields CL, Shields JA, Needle M, Miller D et al. Chemoreduction and local ophthalmic therapy for intraocular retinoblastoma J Clin Oncol 2000;18:12-7.
12. Ellsworth RM. The practical management of retinoblastoma. Trans Am Ophthalmol Soc 1969;67:462-534.
13. Wolff JA, Boesel C, Ellsworth R, Gallie B, Maurer H et al. Extraocular retinoblastoma. New York, 1978 .
14. Cox DR. Regression models on life tables. J R Statist Soc 1972;34B:187-220.
15. Antoneli CBG, Erwenne C, Murini MS, Novaes PE, de Camargo B, Bianchi A. Retinoblastoma (RB) experience in a developing country . Med Pediatr Oncol 1996;27:267. .
16. Erwenne MC. Relação genético-clínica em retinoblastoma . São Paulo: Escola Paulista de Medicina; 1987.
17. Erwenne CM, Camargo B, Antoneli CBG, Marback EF, Novaes PERS. Conservative treatment of retinoblastoma in Brasil . Anais. 10th International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
18. Antoneli CBG, Ribeiro KCB, Erwenne CM, Novaes PERS, Steinhorst F. de Camargo B. Extraocular retinoblastoma. A Ten-year experience . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
19. Shields CL, Shields JA. Recent developments in the management of retinoblastoma. J Pediatr Ophthalmol Strabismus 1999;36:8-18.
20. Meadows AT, Baum E, Fossati-Bellani F, Green D, Jenkin RD, Marsden B et al. Second malignant neoplasms in children: an update from the Late Effects Study Group. J Clin Oncol 1985;3:532-8.
21. Ishi E, Matsuzaki A, Ohnish Y, Kai T, Ueda K. Successful treatment with ranimustine and carboplatin for recurrent intraocular retinoblastoma with vitreous seeding . Proceedings of the Annual Meeting of the American Society of Oncology; 1996.
22. Murphree AL, Villablanca JG, Deegan WF, Sato JK, Malogolowkin M, Fisher A et al. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch Ophthalmol 1996;114:1348-56.
23. Manzitti J, Chantada G, Fandiño A, Schvartzman E. Quimorrecucción com carboplatino y vincristina en el retinoblastoma intraocular. Arch Oftalmol B Aires 1997;72:157-61.
24. Bornfeld N, Schüler A, Bechrakis N, Henze G, Havers W. Preliminary results of primary chemotherapy in retinoblastoma. Klin Padiatr 1997;209:216-21.
25. Bechrakis NE, Bornfeld N, Schueler A, Coupland SE, Henze G, Foerster MH. Clinicopathologic features of retinoblastoma after primary chemoreduction. Arch Ophthalmol 1998;116:887-93.
26. Magramm I, Abramson DH, Ellseworth RM. Optic nerve involvement in retinoblastoma. Ophthalmology 1989;96:217-22.
27. Meadows AT, Friedman D, Hanovar S, Singh AD, Shields J, Shields C. The value of prophylatic chemotherapy in unilateral, enucleated retinoblastoma with factors at high risk for metastase . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
28. Strahlendorf C. Relative frequency and treatment and retinoblastoma in Johannesburg, South Africa: a new vision for developing countries . Med Pediatr Oncol 1997;29:373. .
29. Chan HSL, Calderwood S, Heon E, Fernandez CV, Blaser S, Budning A, Saunders EF, Doyle J, Gallie BL. Combined Intensive multimodality therapy successfully treats extraocular extension of retinoblastoma . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
30. Pratt CB, Rodrigues-Galindo C, Lipson M, Jenkins J, Mercchant T, Kaste S, Wilson MW, Haik BH. Treatment of Extrachoroidal and metastatic retinoblastoma . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
31. Doz F, Khelfaoui F, Mosseri V, Validire P, Quintana E, Michon J et al. The role of chemotherapy in orbital involvement of retinoblastoma. The experience of a single institution with 33 patients. Cancer 1994;74:722-32.
32. Namouni F, Doz F, Tanguy ML, Quintana E, Michon J, Pacquement H et al. High-dose chemotherapy with carboplatin, etoposide and cyclophosphamide followed by a haematopoietic stem cell rescue in patients with high-risk retinoblastoma: a SFOP and SFGM study. Eur J Cancer 1997;33:2368-75.
33. Moll AC, Imhof SM, Bouter LM, Tan KE. Second primary tumors in patients with retinoblastoma. A review of the literature. Ophthalmic Genet 1997;18:27-34.
34. Wong FL, Boice JD, Abramson DH, Tarone RE, Kleinerman RA, Stovall M et al. Cancer incidence after retinoblastoma. Radiation dose and sarcoma risk. Jama 1997;278:1262-7.
35. Harbour JW. Molecular therapy for retinoblastoma . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
36. Barrios CP, Chintagumpala MM, Boniuk M, Hurwitz MY, Hurwitz RL. Suicide gene therapy for retinoblastoma: a phase I clinical trial . In: Thirteenth Biannual Meeting of International Society for Genetic Eye Disease (ISGED) and the Tenth International Symposium on Retinoblastoma. 2001; Fort Lauderdale, Flórida; 2001.
37. Craft AW, Pearson AD. Three decades of chemotherapy for childhood cancer: from cure "at any cost" to cure "at least cost". Cancer Surv 1989; 8:605-29.
Endereço para correspondência
Hospital do Câncer Departamento de Pediatria
Rua Professor Antônio Prudente, 211
São Paulo (SP) - CEP 01509-900
E-mail: [email protected]
Recebido para publicação em 08.04.2002
Aceito para publicação em 27.11.2002
Trabalho realizado no Centro de Tratamento e Pesquisa Hospital do Câncer AC Camargo - Departamento de Oncologia Pediátrica.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. José Wilson Cursino.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}
{kind=link}