Arq. Bras. Oftalmol. 2006; 69 (6): 10.1590/S0004-27492006000600026
Total: 1961
Miguel Gustavo Rosa da Rocha Canêdo1; Luciana Negrão Frota de Almeida3; Ricardo Gonçalves da Silva3; Rafael Negrão Frota de Almeida4; Eurípedes Figueiredo Alessandri5
DOI: 10.1590/S0004-27492006000600026
RESUMO
Os autores relatam um caso de mucopolissacaridose tipo VI em paciente de 19 anos, diagnosticada por meio de exame genético-clínico, demonstrando várias manifestações sistêmicas, incluindo alterações oftalmológicas como: opacidade corneal, aumento da pressão intra-ocular e aumento importante da espessura corneal. Discutem-se os achados característicos sindrômicos e a influência da espessura corneal na alteração da pressão intra-ocular podendo levar a tratamentos antiglaucomatosos desnecessários.
Descritores: Mucopolissacaridose; Glaucoma; Topografia da córnea; Transplante de córnea; Relatos de casos [tipo de publicação]
ABSTRACT
The authors report a case of a 19-year-old patient presenting with type VI mucopolysaccharidosis, diagnosed by genetic-clinical examination, demonstrating several systemic manifestations, including ocular disorders such as: corneal opacity, elevated intra-ocular pressure and increase of corneal thickness. The authors discuss the characteristic syndromic findings and the influence of corneal thickness associated with an increase in intraocular pressure leading to unnecessary antiglaucomatous treatment.
Keywords: Mucopolysaccharidoses; Glaucoma; Corneal topography; Corneal transplantation; Case reports [publication type]
INTRODUÇÃO
As mucopolissacaridoses (MPS) são doenças caracterizadas pelo acúmulo de mucopolissacarídeos ou glicosaminoglicanos (GAGs) em múltiplos órgãos sistêmicos, conseqüente a erros inatos no metabolismo do carbohidrato onde existe a deficiência das enzimas hidrolases lisossomais ácidas em degradarem completamente mucopolissacarídeos como: dermatan sulfato, heparan sulfato, keratan sulfato ou condroitin sulfato(1-5). Mucopolissacarídeos degradados incompletamente e acumulados em vários tecidos levam a manifestações clínicas progressivamente piores tais como: aspereza facial, alterações esqueléticas, cardíacas, cerebrais e oculares(6).
As mucopolissacaridoses são pelo menos oito diferentes síndromes com muitas características em comum, de herança autossômica recessiva, excetuando-se a do tipo II (Síndrome de Hunter) que é ligada ao X(2,7).
O diagnóstico provável de sete tipos clínicos de MPS é baseado em achados clínicos, sendo o diagnóstico definitivo dos subtipos estabelecido por enzimas específicas e correlação destas com o fenótipo(8). Este diagnóstico pode ser obtido por meio de análises bioquímicas de enzimas nas lágrimas, leucócitos, células amnióticas ou fibroblastos cultivados, e, dos níveis urinários elevados de GAGs(3).
Neste relato, objetiva-se, demonstrar como alterações oculares encontradas nestes pacientes, como as relacionadas à espessura corneal, podem levar a conclusões diagnósticas errôneas consequentemente com tratamentos desnecessários e muitas vezes onerosos para os pacientes.
RELATO DE CASO
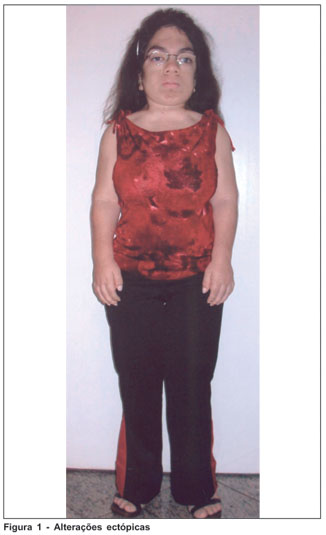
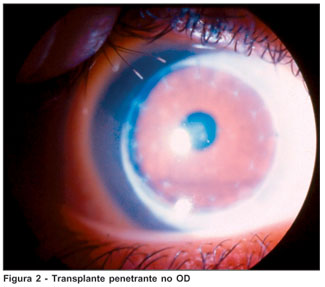
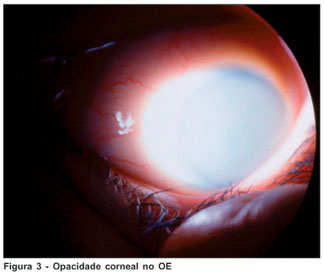
Paciente do sexo feminino, branca, solteira, estudante, 19 anos, natural de Mineiros - GO, portadora de mucopolissacaridose tipo VI (MPS VI), procurou atendimento no Instituto Panamericano da Visão, Goiânia - GO, queixando-se de baixa de acuidade visual no olho esquerdo (OE) sem outros sintomas oculares. Havia nos antecedentes pessoais gerais história de cardiopatia e tratamento sistêmico de mucopolissacaridose. Este iniciado nos EUA com arilsulfatase-B recombinante. Já nos antecedentes familiares, havia história de uma irmã portadora de diabetes juvenil. Os antecedentes pessoais oftalmológicos chamavam atenção na história de transplante penetrante (TP) corneal no olho direito (OD) em setembro de 2003. Em uso de Predmild® no OD e Niolol® no OE. Ao exame refracional a paciente apresentou acuidade visual, com a melhor correção, de 20/30 no OD e 20/200 no OE. À ectoscopia mostrava alterações esqueléticas importantes como nanismo e displasias ósseas características do quadro síndrômico em questão (Figura 1). À biomicroscopia foram observados TP em OD com ótimo aspecto (Figura 2) e opacidade corneal estromal difusa com aspecto de vidro moído associado a opacidades puntiformes finas no estroma profundo (Figura 3). À gonioscopia do OD não foram observados depósitos trabeculares. A pressão intra-ocular, à tonometria de aplanação, com medicação anti-glaucomatosa (somente no OE) era 11 mmHg no OD e 19 mmHG no OE, e, 12 mmHg e 24 mmHg, respectivamente, sem medicação. O exame do fundo de olho do OD mostrou um disco rosado, sem escavação aumentada, enquanto da inviabilidade do OE. A paciente foi submetida à retinografia do OD (caráter de documentação) que teve como resultado a normalidade; ao exame de ecografia ultra-sônica do OE não foram observadas alterações. Foi ainda solicitado Orbscan de ambos os olhos (AO), demonstrando espessura corneal central de 498 µm no OD e 664 µm no OE.
Devido aos resultados obtidos a partir dos exames clínico e complementar, foi decidido pela suspensão da medicação antiglaucomatosa. Há seis meses em acompanhamento, com pressão intra-ocular de 12 mmHg no OD e 24 mmHg no OE à tonometria de aplanação, a paciente foi submetida ao transplante penetrante de córnea do OE em setembro de 2005 com boa evolução pós-operatória.
DISCUSSÃO
Os aspectos oculares mais importantes dizem respeito a infiltrados corneais, degenerações pigmentares retinianas, edema de disco óptico com posterior atrofia óptica e raramente glaucoma. A atrofia óptica é precedida de edema discal óptico, sendo raros na síndrome tipo VI(8).
Alroy et al., demonstraram que os depósitos corneais geralmente estão associados a anomalias esqueléticas, como é o caso em questão(9).
Os mucopolissacarídeos constituem a substância fundamental da córnea (4 a 4,5% do peso da córnea seca), sendo nestas doenças encontrado um excesso de mucopolissacarídeos nos ceratócitos e estroma, e, em alguns casos, no epitélio e endotélio corneais(4,10).
Devido à instabilidade em relação à degradação fisiológica destes mucopolissacarídeos, ocorrem várias alterações de significados variáveis no globo ocular podendo levar à perda da transparência corneal difusa (cerca de 75% dos pacientes com MPS tipo VI apresentam opacificação corneal de intensidade variável) como relatam alguns autores(4) apresentando um aspecto de vidro moído associado a opacidades puntiformes finas no estroma profundo. Relatos de glaucoma já houve, sendo este na verdade um achado questionável, por alguns autores descrito como resultado às alterações trabeculares (depósitos de GAGs)(5,11), e por muitos outros na atualidade interpretado como uma falsa hipertensão ocular devido à alta espessura corneal desses pacientes.
Devemos estar atentos a essa possibilidade de um pseudoglaucoma mascarado por uma córnea espessa, levando a uma superestimação da pressão intra-ocular obtida por tonometria de aplanação desses pacientes, como é o caso em questão aqui relatado.
Este relato sugere uma correlação entre o aumento da pressão intra-ocular e os depósitos de GAGs na córnea, aumentando desta forma a espessura corneal e consequentemente maior seria a sua rigidez, levando assim a aferições superestimadas da pressão intra-ocular à tonometria de aplanação ("pseudo-glaucoma").
É de fundamental importância nestes casos com alterações pressóricas sem alterações correlatas do nervo óptico, uma avaliação criteriosa da espessura corneal, para depois não nos depararmos com um diagnóstico errôneo de glaucoma, e mais, tratamentos clínicos e/ou cirúrgicos desnecessários. Com isso, onerando e mutilando os pacientes devido a um pseudoglaucoma ocasionado pelo aumento da espessura corneal consequente à infiltrados corneais de mucopolissacarídeos.
Concluiu-se então, que o tratamento de escolha para este caso envolveria o transplante penetrante corneal, sem necessidade de tratamento clínico e/ou cirúrgico para o "pseudo-glaucoma". Além disso, trata-se de um paciente com resultado favorável em um dos olhos, com seguimento de dois anos sem recidiva da doença.
Devemos, contudo, continuar acompanhando esse paciente com intuito de obtermos mais informações úteis em relação ao assunto em questão, principalmente, após a realização do transplante penetrante no OE e, com a possibilidade do advento desta nova droga ainda em fase de testes, arilsulfatase-B recombinante, como tratamento sistêmico para as mucopolissacaridoses.
REFERÊNCIAS
1. Bajart AM, Pavilack MA. Corneal manifestations of metabolic diseases. In: Albert DM, Jakobiec FA. Principles and practice of Ophthalmology. Philadelphia: W. B. Saunders; 1994.
2. Neufeld EF, Muenzer J. The mucopolysaccharidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic basics of inherited disease. 6th ed. New York: McGraw-Hill; 1989. p.1565-87.
3. Frangieh GT, Traboulsi EI, Kenyon KR. The mucopolysaccharidosis. In: Gold DH, Weingeist TA, editors. The eye in systemic disease. Philadelphia: WB Lippincott; 1990. p.372-7.
4. Smith RE, Lee JS. The cornea in systemic disease. In: Duane TD. Clinical Ophthalmology. ed. rev. Philadelphia: J.B. Lippincot; 1990.
5. Mullaney P, Awad AH, Millar L. Glaucoma in mucopolysaccharidosis 1-H/S. J Pediatr Ophthalmol Strabismus. 1996;33(2):127-31.
6. Strauch OF, Stypmann J, Reinheckel T, Martinez E, Haverkamp W, Peters C. Cardiac and ocular pathologies in a mouse model of mucopolysaccharidosis type VI. Pediatr Res. 2003;54(5):701-8.
7. Sampaio MW, Kara-José N. Doenças metabólicas. In: Belfort Jr R, Kara-José N. Córnea clínica e cirúrgica. São Paulo: Roca; 1997. p.398-9.
8. Collins ML, Traboulsi EI, Maumenee IH. Optic nerve head swelling and optic atrophy in the systemic mucopolysaccharidosis. Ophthalmology. 1990; 97(11):1445-9.
9. Alroy J, Haskins M, Birk DE. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp Eye Res. 1999; 68(5):523-30.
10. Mollard RJ, Telegan P, Haskins M, Aguirre G. Corneal endothelium in mucopolysaccharide storage disorders. Morphologic studies in animal models. Cornea. 1996;15(1):25-34.
11. Spellacy E, Bankes JL, Crow J, Dourmashkin R, Shah D, Watts RW. Glaucoma in a case of Hurler disease. Br J Ophthalmol. 1980;64(10):773-8. Arq Bras Oftalmol. 2006;69(6):933-5
Endereço para correspondência:
Miguel Canêdo. Al. Couto Magalhães, 830 - Apto. 503 - Ed. Serra da Prata
Goiânia (GO) CEP 74825-040
E-mail: [email protected]
[email protected]
Recebido para publicação em 22.05.2005
Última versão recebida em 31.01.2006
Aprovação em 01.03.2006
Trabalho realizado no Instituto Panamericano da Visão - Goiânia (GO) - Brasil e Hospital da Fundação Banco de Olhos de Goiás - Goiânia (GO) - Brasil.
Os autores declaram não possuírem nenhum interesse financeiro neste estudo.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Rosane da Cruz Ferreira sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.