Arq. Bras. Oftalmol. 2006; 69 (1): 10.1590/S0004-27492006000100018
Total: 3108
Oscar Ewald1; Fernanda Scremin2; Fábio Busch3; Roberto Von Hertwig1
DOI: 10.1590/S0004-27492006000100018
RESUMO
Descreveremos um caso raro de malformações da síndrome de Dandy-Walker e suas alterações oculares. Trata-se de criança do sexo feminino, com idade de 1 ano e 9 meses, que apresentava alterações no sistema nervoso central (síndrome de Dandy-Walker), associado à baixa acuidade visual, megalocórnea (diâmetro de 13 mm no olho direito e 13,5 mm no olho esquerdo), escavação de 0,7 em ambos os olhos (AO) e palidez temporal de papila óptica de AO, pressão intra-ocular de 16 mmHg no olho direito e 14 mmHg no olho esquerdo e diâmetro antero-posterior de 20,55 mm em ambos os olhos.
Descritores: Síndrome de Dandy-Walker; Glaucoma; Malformações do sistema nervoso; Manifestações oculares; Relatos de casos
ABSTRACT
We describe a rare case of Dandy-Walker syndrome malformation and its ocular alterations. A female child, aged 1 year and 9 month, with central nervous system alterations (Dandy-Walker syndrome), associated with low visual acuity, megalocornea (13 mm diameter in right eye and 13.5 mm in the left), fundoscopy with 0.7 optic papilla excavation in both eyes (BE) and temporal paleness in BE, intraocular pressure of 16 mmHg in the right eye and 14 mmHg in the left and anteroposterior diameter of 20.55 mm in both eyes.
Keywords: Dandy-Walker syndrome; Glaucoma; Nervous system malformation; Eye manifestations; Case reports
INTRODUÇÃO
O relato clássico, feito por Dandy & Blackfan em 1914*, revelou casos em autópsias com hidrocefalia severa supratentorial, dilatação cística do quarto ventrículo, vermis pequeno, afastamento dos hemisférios cerebelares, ausência do teto do quarto ventrículo, espessamento e opacificação da pia-aracnóide das cisternas da base do crânio e dilatação do aqueduto(1).
A síndrome de Dandy-Walker (SDW) é uma síndrome não familiar, caracterizada por dilatação cística do quarto ventrículo e por aplasia ou hipotrofia parcial ou total do vermis cerebelar. Geralmente apresenta atresia dos forames de Lushka e Magendie. Em três quartos dos casos ocorrem outras malformações cerebrais como agenesia do corpo caloso, heteropsias, lissencefalia, estenose do aqueduto de Sylvius(2-3). Gardner et al** propuseram que a SDW, juntamente com outras síndromes (Arnold-Chiari, cisto aracnóide de cerebelo e siringomielia), seriam manifestações de uma mesma doença(1).
Alguns estudos mostram uma incidência de aproximadamente 70% de relação entre a SDW e anomalias sistêmicas(1).
Pouco se sabe sobre malformações congênitas das estruturas da fossa posterior, suas alterações genéticas foram mapeadas para o cromossomo 3q(4-5), mas o gene ao certo não localizado, porém sabe-se que a base do processo de desenvolvimento das estruturas da fossa posterior é a natureza para as malformações cerebelares humanas(5). Sabe-se também que as estruturas cerebelares se desenvolvem precocemente no período embrionário até os primeiros anos pós-natais, este acontecimento deixaria o cerebelo vulnerável a um largo espectro de desordens do seu desenvolvimento(6). A patogenia desta síndrome é controvertida, porém a teoria mais aceita é a de que a folha do desenvolvimento dos forames de Lushka e Magendie, durante o quarto mês de vida fetal, leva ao abaulamento cístico do quarto ventrículo. Novas teorias propuseram que a SDW decorreria de uma falha no desenvolvimento no teto do rombencéfalo, tendo este como causa um efeito teratogênico(1). Alguns trabalhos sugerem que o uso de warfarin em longo prazo seria responsável pelo desenvolvimento da SDW em 1-2% dos fetos expostos(7). O que vem reforçar ainda mais esta teoria é que um estudo realizado por Hart et al., mostrou que não há relação entre grau de hidrocefalia e o tamanho do cisto da fossa posterior, atenuação do vermis ou permeabilidade do quarto ventrículo e em alguns casos a hidrocefalia estava ausente(1). A SDW é uma entidade heterogênica de hipoplasia de vermis cerebelar, sendo recentemente identificado um gene associado à ligação com X-HPRT(8), também relacionado com doença de gânglios da base.
Clinicamente pode haver moderado atraso do desenvolvimento psicomotor, microcefalia, hipotonia, mas a sintomatologia predominante se refere à hidrocefalia, geralmente nos dois primeiros anos de vida, esta porém, pode ser ignorada, aparecendo tardiamente (primeira ou segunda década de vida)(2-9). A hidrocefalia se dá pela obstrução dos forames de Lushka e Mangedie(1). Algumas alterações oculares (Tabela) são descritas na SDW, como: coloboma corioretiniano(10), nistag-mo(11-13). Pode haver retardo mental (50%), espasticidade (ao invés de hipotonia), convulsões, vômitos, tudo dependendo do grau da malformação cerebelar(3). Em pacientes com vermis com duas fissuras e conformações praticamente normais, as funções cerebrais são também praticamente normais sem associações com outras malformações. Já em pacientes com severas malformações do cerebelo, vermis com apenas uma ou nenhuma fissura, é comum o retardo mental severo e outras malformações do sistema nervoso central, como agenesia de corpo caloso. Divide-se, assim, a SDW em dois grandes grupos conforme as malformações anteriores para a determinação do prognóstico intelectual(12-13). São relatados na literatura casos de coexistência de grandes hemangiomas cutâneos faciais com a SDW(14).

Outras síndromes onde coexistem malformações cerebrais e oculares são relatadas como síndrome de Neuhauser (MMMM - Megalocornea, macrocephaly, mental and motor retardation), onde são encontrados atrofia cortical, aumento do quarto ventrículo, hipoplasia do corpo caloso. Tudo isto considerado uma variante da SDW, juntamente com megalocórnea(15). Há também a síndrome de Warburg que, além de malformações cerebrais como o cisto de Dandy-Walker, apresenta microftalmo e megalocórnea(16). Esta última também pode ser autossômica dominante ou ligada ao X, bem como associada à síndrome de Marfan. Existem relatos sobre a relação de malformações da fossa posterior com nistagmo, sendo estas chamadas de síndrome cerebelo-oculorenal(11). Outros, sobre síndrome do músculo-olho-cérebro de Santavoure que, além de hidrocefalia, hipotonia, fraqueza, aumento de CPK, há também alta miopia e glaucoma congênito(17). Há também o relato da chamada síndrome PHACE (Posterior fossa malformations, hemangioma, arterial anomalies, coarctation of the aorta and other cardiac defects, and eye abnormalities)(10). Em um estudo entre pacientes com síndrome de Turner, um de seus 23 casos estava presente SDW(18).
Para o diagnóstico há necessidade da ressonância magnética com imagens de boa qualidade da vista axial do vermis cerebelar e imagens em T2(12). Os achados neuroradiológicos são característicos, como a dilatação cística do quarto ventrículo e as alterações no vermis cerebelar(2), além de outras já citadas.
A SDW deve sempre ser acompanhada pelo pediatra, neurocirurgião e um fisioterapeuta. Em caso de convulsões, devem ser usados anticonvulsivantes. Nos casos de hipotonia ou espasticidade, a conduta cabe ao fisioterapeuta(3). O tratamento da hidrocefalia sempre será cirúrgico, através de uma derivação ventrículo-peritoneal(2). Esta poderá ser feita por neuroendoscopia, comunicando o cisto do quarto ventrículo com o sistema ventricular, e este com o peritônio(3,19).
RELATO DE CASO
BSF, 1 ano e 9 meses, sexo feminino, foi referida ao Serviço de Oftalmologia da Clínica de Olhos Roberto Von Hertwig com história de "síndrome de Dandy-Walker" para avaliação oftalmológica (Figura).
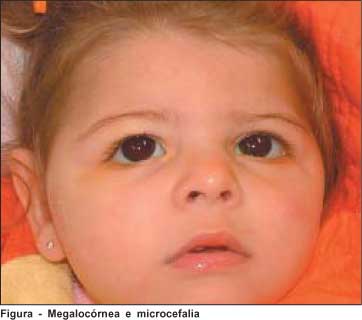
Antecedentes pessoais: Mãe relata o não uso de drogas durante a gestação. Não existindo consangüinidade entre os pais. Criança com ressonância magnética compatível com síndrome de Dandy-Walker, com volumosa coleção na fossa posterior e com ampla comunicação com o quarto ventrículo, associada com sinais de hipoplasia dos hemisférios cerebelares, do vermis e do tronco cerebelar. Cariótipo não realizado. Mãe relata que a criança tem microcefalia, segundo o pediatra; também apresenta história de nistagmo.
Exame oftalmológico: Criança fixa luz, mas não segue luz nem objetos pequenos. Ortofórica e ortotrópica e ausência de nistagmo. Na biomicroscopia, apresenta megalocórnea, com córnea de 13 mm no olho direito e 13,5 mm no olho esquerdo. A refração estática sob cicloplegia do olho direito é +5,00 dioptrias esféricas (DE) e -3,50 dioptrias cilíndricas (DC) a 180º e do olho esquerdo é +5,00DE e -4,00DC a 180º. Fundoscopia com escavação de 0,7 no disco óptico de AO e palidez temporal dos discos em AO, sendo escavação maior que a palidez. Pressão intra-ocular, sem sedação, de 16 mmHg no olho direito e 14 mmHg no olho esquerdo. Ecobiometria demonstrando diâmetro antero-posterior do olho de 20,55 mm em ambos os olhos. Sem fotofobia, lacrimejamento ou estrias de Haab (ou outra alteração da córnea).
Foram então prescritas lentes para correção da visão com +1,50DE» -1,50DC a 180º no olho direito e +1,50DE» -2,00DC a 180º e encaminhada para estimulação visual.
DISCUSSÃO
O fato de a paciente ter escavação de 0,7 no disco óptico de AO com palidez temporal em AO, aliada à existência de megalocórnea, pode sugerir glaucoma congênito.
Porém a paciente é hipermétrope, com diâmetro antero-posterior normal para a idade, pressão intra-ocular, sem sedação, não aumentada, sem fotofobia, lacrimejamento ou estrias de Haab (ou outra alteração da córnea), não sugerindo o diagnóstico de glaucoma congênito.
Existem trabalhos que citam a existência de megalocórnea com malformações neurológicas e variantes da SDW(15-16).
A hipótese diagnóstica mais provável é que, por se tratar de uma síndrome com malformações neurológicas(2-3), esta seja a origem do comprometimento do disco óptico.
Foram prescritas lentes corretoras para que a criança pudesse ser visualmente estimulada, evitando ambliopia.
Outras alterações comuns na síndrome de Dandy-Walker, como coloboma corioretiniano(10) e nistagmo(3,11), não foram encontradas no momento.
Foi dada orientação aos pais para nova avaliação em dois meses.
REFERÊNCIAS
1. Diament A. Neurologia infantil 3a ed. São Paulo: Atheneu; 1996.
2. Rosenberg S. Neuropediatria. São Paulo: Sarvier; 1995.
3. Yilmaz MA. The site on Dandy Walker Syndrome [homepage on the Internet]. [cited 2005 Aug 29]. Available from: http://www.geocities.com/murat_yil/dandy.html
4. Parisi MA, Dobyns WB. Human malformations of the midbrain and hindbrain: review and proposed classification scheme. Mol Genet Metab. 2003; 80(1-2):36-53.
5. Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003;80(1-2):54-65.
6. Ten Donkelaar HJ, Lammens M, Wesseling P, Thijssen HO, Renier WO. Development and developmental disorders of the human cerebellum. J Neurol. 2003;250(9):1025-36.
7. Koren G, Pastuszak A, Ito S. Drugs in pregnancy. N Engl J Med. 1998;338(16): 1128-37.
8. Daufenbach DR, Ruttum MS, Pulido JS, Keech RV. Chorioretinal colobomas in a pediatric population. Ophthalmology. 1998;105(8):1455-8. Comment in: Ophthalmology. 1999;106(4):645-6.
9. Niesen CE. Malformations of the posterior fossa: current perspectives. Semin Pediatr Neurol. 2002;9(4):320-34.
10. Coats DK, Paysse EA, Levy ML. PHACE: a neurocutaneous syndrome with important ophthalmologic implications: case report and literature review. Ophthalmology. 1999;106(9):1739-41.
11. Kumandas S, Akcakus M, Coskun A, Gumus H. Joubert syndrome: review and report of seven new cases. Eur J Neurol. 2004;11(8):505-10.
12. Klein O, Pierre-Kahn A, Boddaert N, Parisot D, Brunelle F. Dandy-Walker malformation: prenatal diagnosis and prognosis. Childs Nerv Syst. 2003;19(78): 484-9.
13. Boddaert N, Klein O, Ferguson N, Sonigo P, Parisot D, Hertz-Pannier L, et al. Intellectual prognosis of the Dandy-Walker malformation in children: the importance of vermian lobulation. Neuroradiology. 2003;45(5):320-4.
14. Reese V, , Paller AS, Esterly NB, Ferriero D, Levy ML, et al. Association of facial hemangiomas with Dandy-Walker and other posterior fossa malformations. J Pediatr 1993;122(3):379-84.
15. Balci S, Teksam O, Gedik S. Megalocornea, macrocephaly, mental and motor retardation: MMMM syndrome (Neuhauser syndrome) in two sisters with hypoplastic corpus callosum. Turk J Pediatr. 2002;44(3):274-7.
16. Pagon RA, Clarren SK, Milam DF Jr, Hendrickson AE. Autosomal recessive eye and brain anomalies: Warburg syndrome. J Pediatr. 1983;102(4):542-6.
17. Golden JA. Cell migration and cerebral cortical development. Neuropathol Appl Neurobiol. 2001;27(1):22-8.
18. Ruibal Francisco JL, Sánchez Buron P, Piñero Martinez E, Bueno Lozano G. [Turner's syndrome. Relationship between the karyotypes and malformations and associated diseases in 23 patients]. An Esp Pediatr. 1997;47(2):167-71. Spanish.
19. Kawaguchi T, Jokura H, Kusaka Y, Shirane R, Yoshimoto T. Intraoperative direct neuroendoscopic observation of the aqueduct in Dandy-Walker malformation. Acta Neurochir (Wien). 2003;145(1):63-7.
Endereço para correspondência:
Oscar Ewald
Rua Procópio Domingos Alexandre, 90
Blumenau (SC) CEP 89037-360
E-mail: [email protected]
[email protected]
Recebido para publicação em 04.05.2005
Versão revisada recebida em 04.10.2005
Aprovação em 29.10.2005
Trabalho realizado na Clínica de Olhos Roberto Von Hertwig - Rua Itajaí, 417 - Blumenau (SC) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Simone Finzi sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.
* Dandy & Blackfan apud(1)
** Gardner et al. apud(1)

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.