Arq. Bras. Oftalmol. 2000; 63 (3): 10.1590/S0004-27492000000300007
Total: 2568
Fábio Henrique Cacho Casanova1; Rodrigo L. Meirelles1; Marcelo Tojar2; Maria Cristina Martins3; Moacir P. Rigueiro4; Denise de Freitas2
DOI: 10.1590/S0004-27492000000300007
RESUMO
Contexto: A vasculite leucocitoclástica é uma doença que acomete pequenos vasos mais comumente associada a distúrbios do tecido conectivo, caracterizada clinicamente pela presença de lesões purpúricas palpáveis. Manifestações oftalmológicas são raras. Objetivo: Descrever quadro clínico de paciente portador de vasculite leucocitoclástica como manifestação incomum de padrão granulomatoso do erythema elevatum diutinum (EED). Relato de Caso: Paciente de 64 anos, masculino, apresentando baixa de acuidade visual e aparecimento de lesões nodulares em mãos. Ao exame oftalmológico, evidenciou-se afinamento corneano superior bilateral com perfuração corneana do olho esquerdo e espessamento da conjuntiva limbar em ambos os olhos, associada a intensa atividade inflamatória. Exame anatomopatológico da conjuntiva revelou vasculite granulomatosa com intenso exsudato neutrofílico, células gigantes multinucleadas, além de proliferação fibroblástica. Exame anatomopatológico de biopsia da pele evidenciou alterações semelhantes compatíveis com EED, porém não foi visto célula gigante sugerindo lesão granulomatosa. Conclusão: Ceratólise auto-imune secundária a vasculite leucocitoclástica cutânea (EED) não havia sido descrito na literatura, tampouco encontramos referências sobre a reação granulomatosa (encontrada na conjuntiva) em EED.
Descritores: Vasculite leucocitoclástica; Ceratólise; Erythema elevatum diutinum; Perfuração ocular
ABSTRACT
Purpose: Leucocytoclastic vasculitis is an immune complex-mediated, small vessel disease, that is characterized clinically by the presence of palpable purpuric lesions, most often in association with rheumatic diseases. Ocular manifestations are rare. We describe a patient with an unusual granulomatous pattern of EED associated with autoimmune keratolysis. Case Report: A 64-year-old man with decreased visual acuity and nodular lesions in both hands. Ocular exami-nation presented bilateral superior corneal melting with perfuration in left eye and conjunctival thickness in both eyes, in association with severe inflammatory reaction. His-topathologic examination of the conjunctiva revealed gra-nulomatous vasculitis with neutrophilic infiltrate, giant cells and fibroblastic proliferation. A punch biopsy from skin showed similar characteristics suggesting EED, however there was no giant cell. Conclusion: To our knowledge, autoimmune keratolysis secondary to cutaneous leucocytoclastic vasculitis (EED) has not previously been described, neither did we find a des-cription about granulomatous reaction (from conjunctiva) in EED.
Keywords: Leucocytoclastic vasculitis; Keratolysis; Ery-thema elevatum diutinum; Ocular perfuration
INTRODUÇÃO
A vasculite leucocitoclástica (VLCC), conhecida também como vasculite alérgica, angiíte por hipersensibilidade e vasculite necrotizante, é uma doença que acomete pequenos vasos (vênulas pós-capilares) mediada por imunocomplexos. É mais comumente associada a distúrbios do tecido conectivo, caracterizada clinicamente pela presença de lesões cutâneas purpúricas palpáveis mais freqüentemente localizadas nas extremidades inferiores 1. Aproximadamente 50% dos pacientes têm envolvimento de pelo menos um órgão além da pele 2. Os órgãos mais acometidos são rins, articulações, pulmões, cérebro e trato gastrointestinal 3. Os achados laboratoriais mais comuns incluem alterações na velocidade de hemossedimentação (VHS) e urinálise, leucocitose leve com ou sem eosinofilia, fator reumatóide (FR) positivo e fator antinuclear (FAN) positivo 4. Manifestações oftalmológicas da VLCC são raras, porém há relatos sugerindo associação com uveíte, retinite multifocal, episclerite, pseudotumor cerebral, e atrofia óptica 5.
Erythema elevatum diutinum (EED) é uma afecção cutânea crônica, rara, caracterizada clinicamente por pápulas amarelo-avermelhadas, placas e nódulos. Histopatologicamente, o EED se apresenta inicialmente como uma VLCC na qual os leucócitos polimorfonucleares são as células inflamatórias predominantes. Em lesões avançadas, o infiltrado inflamatório é substituído por tecido de granulação, fibrose e depósito de material lipídico 6, 7.
OBJETIVO
Descrever o quadro clínico de um paciente portador de VLCC como apresentação incomum do EED de padrão granulomatoso a fim de alertar a possibilidade da doença.
RELATO DE CASO
Paciente de 64 anos, sexo masculino, queixando-se de baixa da acuidade visual em ambos os olhos há 5 meses. Refere o aparecimento de lesões nodulares indolores em mãos, face externa do antebraço e membros inferiores, dor nas articulações interfalangeanas proximais e distais com presença de rigidez matinal (>1h). Vinha fazendo uso de prednisona 60 mg/d há 18meses. Referia ainda uma perda de 20kg nos últimos 2 anos.
Ao exame oftalmológico, apresentava acuidade visual com melhor correção de 0,3 no olho direito (OD) e movimento de mão no olho esquerdo (OE). A pressão intraocular era de 16mmHg em ambos os olhos. À lâmpada de fenda, apresentava um afinamento corneano superior com bordas sobrelevadas, pannus superior, espessamento conjuntival próximo à região inferior da córnea em OD. No olho esquerdo, observava-se hiperemia conjuntival, atalamia, afinamento corneano superior associado à perfuração, edema de córnea (+++), pannus e espessamento conjuntival em região superior, presença de infiltrado estromal com espículas na região inferior da córnea (Fig. 1).
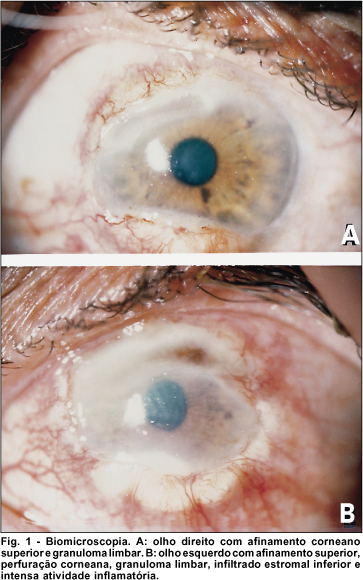
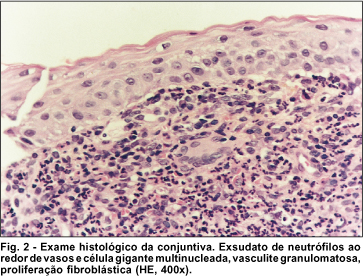
Paciente foi submetido a ressecção da lesão conjuntival do OE e aplicação de tecido adesivo (cianoacrilato) sobre a perfuração. O exame anatomopatológico da lesão conjuntival revelou vasculite granulomatosa no córion superficial, com intenso exsudato de neutrófilos e discreta leucocitoclase (cariorexis do núcleo dos leucócitos), havendo células gigantes multinucleadas ao redor, além da proliferação fibroblástica (Fig.2).
Foi iniciado anti-inflamatório não-hormonal e antibiótico tópicos. Cerca de uma semana após cirurgia, paciente evoluiu com piora das lesões cutâneas, surgindo púrpuras pápulo-eritematosas e petéquias nos joelhos, dorso dos pés e mãos, além de alguns nódulos justa-articulares no dorso das mãos, tendo sido internado e iniciado ciclofosfamida 100mg/d. Durante internação, apresentou duas paradas cardíacas e desenvolveu abscesso cervical e mediastinal por Staphylococcus aureus.
Exames laboratoriais incluíram hemograma com plaquetas, urinálise, creatinina sérica, crioglobulinas, FR, FAN, dosagem de complemento (CH50, C2 hemolítico), DNA nativo, SM, SSA (Ro), SSB (La), anticorpo anticardiolipina (MPL e GPL), ANCA, transaminases, eletroforese de proteínas, estando todos dentro dos padrões de normalidade. Apenas VHS, gama GT, PCR e contagem de leucócitos encontravam-se aumentados. Testes imunológicos para investigação de doenças do tecido conectivo foram realizados, porém não revelaram possível associação com as mesmas.
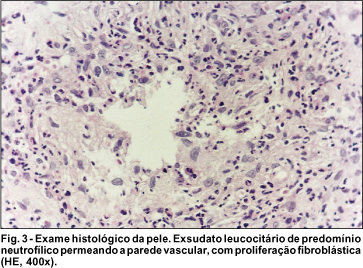
Realizada biópsia de pele em região dorsal da mão tendo sido diagnosticado quadro de VLCC, sugerindo EED. Observou-se a presença de edema e necrose de células endoteliais; invasão da parede vascular por leucócitos, particularmente polimorfonucleares; cariorexis do núcleo dos leucócitos (leucocitoclase) e depósito de "poeira nuclear" sobre os vasos, com proliferação fibroblástica perivascular (Fig. 3).
Após 54 dias, paciente evoluiu com melhora do quadro sistêmico tendo recebido alta hospitalar e suspenso o uso de ciclofosfamida e mantida a imunossupressão com prednisona 20mg/d. Paciente evoluiu de forma satisfatória, apresentando no momento quadro ocular estável, sem sinais de descompensação corneana. Apresenta acuidade visual do olho direito de 20/40 com melhor correção e movimento de mão em OE.
DISCUSSÃO
A vasculite leucocitoclástica (VLCC), em sua apresentação clínica mais típica, é caracterizada pelo surgimento de lesões purpúricas palpáveis em 1 a 2 dias nas extremidades inferiores, embora possam ocorrer em qualquer parte do corpo. Lesões precoces freqüentemente não são palpáveis. Tornam-se mais evidentes à palpação ao aumentarem, podendo coalescer, e alcançar até 1 cm de diâmetro. Geralmente aparecem de forma abrupta e desaparecem em poucos dias 1, 8.
Sintomas constitucionais incluem febre e artralgias na maioria dos pacientes. Pode-se ainda evidenciar sinovite palpável, envolvimento renal (proteinúria, hematúria, e ocasionalmente, insuficiência), dor abdominal e sangramento gastrointestinal, infiltrados pulmonares, acometimento neurológico (cefaléia, diplopia), anemia normocrômica, aumento VHS, leucocitose e eosinofilia 1, 2, 4, 8.
Para se ter o diagnóstico de VLCC, devem ser preenchidos pelo menos 3 dos seguintes critérios: a) idade de início da doença maior que 16 anos; b) uso de medicação no início do quadro; c) púrpura palpável (não relacionada a trombocitopenia); d) exantema maculopapular; e) biópsia incluindo arteríola e vênula (granulócitos em região perivascular ou extravascular) 9.
Zeek et al. subclassificaram as vasculites baseados em critérios histopatológicos, incluindo o tamanho e tipo de vasos afetados e o tipo de inflamação. Tem-se, portanto, vasculites que afetam artérias e/ou veias com inflamação aguda ou crônica (arterite temporal, arterite de Takayasu, poliarterite nodosa) ou inflamação granulomatosa (granulomatose de Wegener); além daquela que acomete vênulas ou arteríolas (VLCC) 10, 11.
Recentemente, tem-se adotado uma nova subdivisão baseada na apresentação da doença, a saber: VLCC predominantemente sistêmica, predominantemente cutânea ou ainda uma síndrome mista de acometimento sistêmico e cutâneo 12. Embora qualquer órgão possa ser afetado por este tipo de vasculite, o comprometimento cutâneo geralmente domina o quadro clínico, enquanto o comprometimento extracutâneo costuma ser bem menos grave que aquele observado nas vasculites sistêmicas 13.
O exame histopatológico mostra o envolvimento de vênulas pós-capilares com necrose fibrinóide da parede vascular e edema de células endoteliais. Leucócitos polimorfonucleares estão presentes na parede vascular e no tecido conectivo perivascular, acompanhado por leucocitoclasia (cariorexis do núcleo do leucócito) e depósito de "poeira nuclear". Outras alterações incluem hemorragia, trombose e, em lesões tardias, proliferação fibroblástica. Pelo menos 2 das 4 primeiras alterações são necessárias para o diagnóstico histológico 1, 8, 12, 13. A fim de se obter histologia específica, a biópsia deve ser realizada em lesões recentes (menos de 24h) 1. Imunofluorescência direta de uma lesão com menos de 8h freqüentemente revela depósito granular de IgG, IgM, IgA e C3 na parede vascular. Em lesões mais antigas, geralmente as imunoglobulinas já desapareceram 14. O depósito de imunoglobulinas sugere fortemente uma vasculite mediada por imunocomplexos 15.
De acordo com a literatura, o percentual de pacientes com VLCC no qual uma condição associada foi identificada varia de 39 a 61% 15. Geralmente, está associada a doenças do tecido conectivo, infecções e medicamentos. Dentre os agentes infecciosos, os mais reconhecidos são Streptococcus e o vírus da hepatite B. Os medicamentos mais envolvidos são antibióticos (especialmente penicilina e sulfonamida), tiazídicos e anti-inflamatórios não-hormonais. Crioglobulinemia também pode estar associada 1. Recentes relatos descrevem a associação com hepatite C, Meningococcus, Pseudomonas e Campylobacter 15. Pode estar associada a certas neoplasias malignas, em particular neoplasias linfóides ou reticuloendoteliais. De particular interesse é a associação entre leucemia de células pilosas e poliarterite nodosa clássica 13. Uma vasculite leucocitoclástica que afeta predominantemente a pele, com comprometimento ocasional de outros sistemas orgânicos, pode constituir um componente secundário de muitas outras doenças. Essas doenças incluem endocardite bacteriana subaguda, infecção por vírus de Epstein-Barr, hepatite crônica ativa, colite ulcerativa, deficiências congênitas de diversos componentes do complemento, fibrose retroperitoneal e cirrose biliar primária. Foi ainda relatada associação com deficiência de alfa1-antitripsina, cirurgia de derivação intestinal e policondrite recidivante 13. No caso em questão, não foram identificadas patologias associadas.
Há relatos de associação de níveis de anticorpo anticardiolipina positivo e VLCC 16, bem como HLA-DR4 associado com manifestações extra-articulares (como vasculite) em pacientes com artrite reumatóide 17.
Pode-se observar dois padrões distintos de infiltrado celular, um predominantemente mononuclear e outro neutrofílico. Zax et al. têm demonstrado uma seqüência temporal onde o infiltrado inflamatório visto nas lesões vasculíticas é predominantemente neutrofílico, e gradualmente evolui para um padrão predominantemente mononuclear 18. Há algumas condições (como granuloma faciale e EED) caracterizadas histologicamente por VLCC que têm uma evolução crônica e não seguem esta seqüência histológica 19.
Foram observadas, no paciente em questão, lesões papulonodulares e placas que associadas aos achados histopatológicos, sugerem uma síndrome de VLCC cutânea conhecida como EED. É um distúrbio cutâneo crônico raro de etiologia desconhecida, caracterizado por lesões persistentes simetricamente distribuídas sobre as superfícies extensoras das extremidades (mãos, joelhos e cotovelos) 12, 20. Podem iniciar-se como pápulas pequenas que coalescem para formar placas anulares ou nódulos. São geralmente firmes e indolores, e podem ser eritematosas, violáceas ou amareladas. Em raros casos, as lesões podem ulcerar ou vesicular 12, 20.
No EED, histologicamente, as lesões recentes e a grande maioria das tardias caracterizam-se pela VLCC associadas a infiltrado denso composto primariamente de neutrófilos e leucocitoclasia, e ocasionalmente monócitos. Algumas lesões tardias apresentam tecido de granulação e proliferação fibroblástica 6, 7, 20, 21. Devido à evolução crônica, desenvolve-se um padrão histológico mais complexo caracterizado por uma mistura de linfócitos, plasmócitos e macrófagos, além de alterações hemorrágicas e fibroblásticas 21.
Nosso paciente apresentava quadro clínico e histológico típico de EED, exceto pelo importante achado na microscopia óptica de uma célula gigante multinucleada após biópsia conjuntival. Essa apresentação incomum do EED com padrão granulomatoso e envolvimento ocular não havia sido descrita anteriormente. Há apenas uma breve citação de raras células gigantes multinucleadas em uma lesão cutânea antiga, porém sem acometimento ocular 22.
A etiologia do EED é desconhecida. Tem sido proposto que a VLCC seja iniciada por depósito de imunocomplexos com ativação de complemento 6, 20, 23. Entretanto, a presença de células de Langerhans descrita por alguns autores sugere um possível papel relacionado a reações mediadas por células na patogênese do EED 22, 24-26. Além disso, a presença de granuloma, como no nosso caso, sugere uma reação de hipersensibilidade tardia mediada por células T 8.
No EED o envolvimento sistêmico é praticamente ausente, porém associações com gamopatias monoclonais 7, 20, infecções recorrentes por Streptococcus 20, doenças do colágeno 7, infecção por HIV 24, 27, policondrite recidivante 28, granulomatose de Wegener 23 e artrite reumatóide 29 têm sido descritas.
Extensa avaliação clínica e laboratorial em nosso paciente não revelou evidência de doenças do tecido conectivo ou outras patologias associadas. Há poucos casos na literatura sobre EED relacionado à doença corneana periférica. Takiwaki et al. descreveram três casos de ceratite ulcerativa periférica associada a EED e altos títulos de fator reumatóide 29. Nosso paciente apresenta fator reumatóide negativo. Shimazaki et al. descreveram um caso de degeneração marginal de Terrien associada a EED, mas nesse caso o epitélio corneano está íntegro 30. Wilkinson et al. relataram rapidamente um caso com úlcera corneana recorrente 21.
Nós descrevemos um paciente portador de EED padrão granulomatoso associado à ceratólise autoimune. As lesões oculares e cutâneas responderam, com sucesso, ao tratamento com corticosteróide e ciclofosfamida.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Soter NA. Cutaneous necrotizing venulitis. In: Fitzpatrick TB, Eisen AZ, Wolff K, Freedberg IM, Austen KF, eds. Dermatology in general medicine, vol.1, 4th ed. New York: McGraw-Hill, 1993;1501-10.
2. Sams WM Jr. Small vessel vasculitis. In: Sams WM Jr, Lynch PJ, eds. Principles and practice of dermatology. New York: Churchill Livingstone, 1990;523-31.
3. Ramsay C, Fry L. Allergic vasculitis: clinical and histological features and incidence of renal involvement. Br J Dermatol 1969;81:96-102.
4. Sanchez NP, Van Hale HM, Su WPD. Clinical and histopathologic spectrum of necrotizing vasculitis: report of findings in 101cases. Arch Dermatol 1985;121:220-4.
5. Tsai JC, Forster DJ, Ober RR, Rao NA. Panuveitis and multifocal retinitis in a patient with leukocytoclastic vasculitis. Br J Ophthalmol 1993;77:318-20.
6. Sangueza OP, Pilcher B, Sangueza JM. Erythema elevatum diutinum: a clinicopathological study of eight cases. Am J Dermatopathol 1997;19:214-22.
7. Yiannias JA, El-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol 1992;26:38-44.
8. Valente RM, Hall S, O'Duffy JD, Conn DL. Vasculitis and related disorders. In: Kelley WN, Harris ED, Ruddy S, Sledge CB, eds. Textbook of Rheumatology, vol.2, 5th ed. Philadelphia: WB Saunders, 1997:1079-122.
9. Calabrese LH, Michel BA, Bloch DA, Arend WP, Edworthy SM, Fauci AS, Fries JF, Hunder GG, Leavitt RY, Lie JT, Lightfoot RW Jr, Masi AT, McShane DJ, Mills JA, Stevens MB, Wallace SL, Zvaifler NJ. The American College of Rheumatology 1990 criteria for the classification of hypersensitivity vasculitis. Arthritis Rheum 1990;33:1108-13.
10. Zeek PM. Periarteritis nodosa: a critical review. Am J Clin Pathol 1952;22:777-90.
11. Zeek PM. Periarteritis nodosa and other forms of necrotizing angiitis. N Engl J Med 1953;248:764-72.
12. Gammon R. Leucocytoclastic vasculitis. Clin Rheum Dis 1982;8:397-413.
13. Fauci AS. The vasculitis syndromes. In: Fauci AS, Braunwald E, Isselbacher KJ, Wilson JD, Martin JB, Kasper DL, Hauser SL, Longo DL, eds. Harrison's principles of internal medicine, 14th ed. New York: McGraw-Hill, 1998:1910-22.
14. Boom BW, Mommaes AM, Vermeer BJ. Presence and interpretation of vascular immune deposits in human skin: the value of direct immunofluorescence. J Dermatol Science 1992;3:26-34.
15. Gyselbrecht L, De Keyser F, Ongenae K, Naeyaert JM, Praet M, Veys EM. Etiological factors and underlying conditions in patients with leucocytoclastic vasculitis. Clin Exp Rheumatol 1996;14:665-8.
16. Katayama I, Masuzawa M, Nishioka K, Nishiyama S. Anticardiolipin antibody in Henoch-Schonlein purpura and related vascular disorders. Arch Dermatol Res 1989;281:296-8.
17. Westedt ML, Breedveld FC, Schreuder GM, D'Amaro J, Cats A, De Vries RRP. Immunogenetic heterogeneity of rheumatoid arthritis. Ann Rheum Dis 1986;45:534-8.
18. Zax RH, Hodge SJ, Callen JP. Cutaneous leukocytoclastic vasculitis: serial histopathologic evaluation demonstrates the dynamic nature of the infiltrate. Arch Dermatol 1990;126:69-72.
19. Smoller BR, McNutt NS, Contreras F. The natural history of vasculitis: what the histology tells us about pathogenesis. Arch Dermatol 1990;126:84-9.
20. Katz SI, Gallin JI, Hertz KC, Fauci AS, Lawley TJ. Erythema elevatum diutinum: skin and systemic manifestations, immunologic studies and successful treatment with dapsone. Medicine 1977;56:443-55.
21. Wilkinson SM, English JSC, Smith NP, Wilson-Jones E, Winkelmann RK. Erythema elevatum diutinum: a clinicopathological study. Clin Exp Dermatol 1992;17:87-93.
22. Lee AY, Nakagawa H, Nogita T, Ishibashi Y. Erythema elevatum diutinum: an ultrastructural case study. J Cutan Pathol 1989;16:211-7.
23. Kavanagh GM, Colaco CB, Bradfield JW, Archer CB. Erythema elevatum diutinum associated with Wegener's granulomatosis and IgA paraproteinemia. J Am Acad Dermatol 1993;28:846-9.
24. Da Cunha Bang F, Weismann K, Ralfkiaer E, Pallesen G, Wantzin GL. Erythema elevatum diutinum and pre-AIDS. Acta Derm Venereol (Stockh) 1986;66:272-4.
25. Kanitakis J, Cozzani E, Lyonnet S, Thivolet J. Ultrastructural study of chronic lesions of erythema elevatum diutinum: "Extracellular cholesterosis" is a misnomer. J Am Acad Dermatol 1993;29:363-7.
26. Porneuf M, Duterque M, Sotto A, Jourdan J. Unusual erythema elevatum diutinum with fibrohistiocytic proliferation. Br J Dermatol 1996;134:1131-4.
27. Dronda F, Gonzalez-Lopez A, Lecona M, Barros C. Erythema elevatum diutinum in human immunodeficiency virus-infected patients: report of a case and review of the literature. Clin Exp Dermatol 1996;21:222-5.
28. Bernard P, Bedane C, Delrous JL, Catanzano G, Bonnetblanc JM. Erythema elevatum diutinum in a patient with relapsing polychondritis. J Am Acad Dermatol 1992;26:312-5.
29. Takiwaki H, Kubo Y, Tsuda H, Arase S, Shiota H. Peripheral ulcerative keratitis associated with erythema elevatum diutinum and a positive rheumatoid factor: a report of three cases. Br J Dermatol 1998;138:893-7.
30. Shimazaki J, Yang HY, Shimmura S, Tsubota K. Terrien's marginal degeneration associated with erythema elevatum diutinum. Cornea 1998;17:342-4.
Universidade Federal de São Paulo ¾ Escola Paulista de Medicina.
(1) Médico Residente do Departamento de Oftalmologia da Universidade Federal de São Paulo ¾ Escola Paulista de Medicina.
(2) Setor de Doenças Externas e Córnea do Departamento de Oftalmologia da UNIFESP ¾ EPM.
(3) Chefe do Setor de Patologia Ocular do Departamento de Oftalmologia da UNIFESP ¾ EPM
(4) Departamento de Patologia da UNIFESP ¾ EPM.
(5) Chefe do Setor de Doenças Externas e Córnea do Departamento de Oftalmologia da UNIFESP ¾ EPM.
Endereço para correspondência: Fábio Casanova. Rua Loefgreen, 1654/81 - Vl. Clementino - São Paulo (SP) CEP 04040-002. Fone: (011) 571-7879/5085-2000. Fax: (011) 573-4002. e-mail: [email protected]

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.