Arq. Bras. Oftalmol. 2004; 67 (2): 10.1590/S0004-27492004000200027
Total: 2207
Vanderson Glerian Dias1; David Ribeiro de Mendonça Filho3; Marcia Abelin Vargas3; Flávia Fauaz Gonçalves1; Edmilson Gigante4; Fátima Jesus Passador Valério5
DOI: 10.1590/S0004-27492004000200027
RESUMO
Os autores apresentam um caso de síndrome de Dubowitz em criança de 7 anos de idade, diagnosticada por meio de exame genético-clínico, demonstrando manifestações nos mais variados sistemas, incluindo alterações oftalmológicas como: epicanto e exotropia com pequeno componente vertical incomitante associado e discreta variação alfabética em A. Discute-se a importância do oftalmologista no diagnóstico e encaminhamento precoces de pacientes com esta síndrome e propõe-se uma forma mais adequada de acompanhamento desses pacientes.
Descritores: Oftalmopatias; Anormalidades múltiplas; Aberrações cromossômicas; Transtornos do crescimento; Síndrome; Relato de caso
ABSTRACT
The authors present a 7-year-old child with Dubowitz syndrome, diagnosed by means of a clinical-genetic examination, showing manifestations in various systems, including ophthalmologic alterations such as: epicanthal folds and exotropia associated with a small incomitant vertical component and discret alphabetic A variation. We discuss the importance of the ophthalmologist in the early diagnosis and early indication for treatment of the patients with this syndrome and propose the most adequate follow-up of these patients.
Keywords: Eye diseases; Multiple abnormalities; Chromossome aberrations; Growth disorders; Syndrome; Case report
INTRODUÇÃO
Esta síndrome foi primeiramente descrita em 1965 pelo Dr. Victor Dubowitz(1) e só mais tarde, em 1971, designada como síndrome de Dubowitz por Gorlin & Opitz ambos da University Medical School of Wisconsin(2).
Há um grande espectro de fenótipos podendo afetar os sistemas imune, hematológico, neurológico, urológico, cardiovascular, músculo-esquelético, digestivo, as regiões cutâneas, os dentes e os olhos. O quadro clínico ocular inclui: estrabismo, ptose palpebral, inflamações crônicas dos canais lacrimais, blefarofimoses, fenda palpebral diminuída, hipertelorismo, telecanto, epicanto, alterações do fundo de olho (vasos tortuosos, malformações do disco óptico e retina, alteração de pigmentação)(3-6). Vários autores descreveram associações importantes com esta síndrome, como, casos apresentando coloboma de íris(7); associação com glaucoma(8), com paralisia do nervo oculomotor direito(9).
É uma síndrome herdada de forma autossômica recessiva e não há teste laboratorial para diagnóstico, sendo este feito clinicamente, após exclusão de outras síndromes genéticas. Não se sabe ainda a localização do gene nem sua patogenia, sendo sugerido como fator responsável, a ação intracelular de genótipo mutante em várias ocasiões durante o período pré e pós-natal do desenvolvimento(7).
Seu quadro clínico mais comum é: retardo de crescimento pré e pós-natal, microcefalia, pequena estatura e alterações faciais e físicas características. Tipicamente os pacientes nascem com peso normal e desenvolvem diminuição severa do ganho de peso pós-natal, porém todos os pacientes apresentam, até o momento, estatura menor com estrutura corporal normalmente proporcional(7).
Por muitos anos esta síndrome foi considerada rara, porém sabe-se hoje que, na verdade, simplesmente muitos pacientes não são diagnosticados adequadamente, seus tratamentos e cuidados multidisciplinares não realizados prontamente deixando suas situações clínicas especiais para segundo plano. Objetiva-se, com este relato, orientar os oftalmologistas para que possam encaminhar adequadamente os pacientes suspeitos e para que saibam diagnosticar como sindrômicas, as alterações oculares da síndrome.
RELATO DO CASO
A.J.O., masculino, branco, sete anos, natural e procedente de Santo Anastácio – SP. Nasceu de parto normal a termo, sem intercorrências, pesando 2.200 g, PIG (pequeno para idade gestacional), estatura de 46 cm, Apgar de 9 e 10. Mãe relata ter feito pré-natal adequadamente e nega qualquer infecção, além de afirmar não ter usado bebida alcoólica, cigarro ou drogas ilícitas durante a gravidez.
Avaliado pelo serviço de Pediatria do Hospital Universitário (HU) da Faculdade de Medicina de Presidente Prudente – SP (FAMEPP) da Universidade do Oeste Paulista (UNOESTE) em julho de 2000, o paciente apresentava irritação, agitação psicomotora, déficit de crescimento e estrabismo divergente. O menor foi encaminhado para avaliação endocrinológica, sendo demonstrado o mesmo quadro clínico anteriormente descrito, critério de Tanner G1-P1, estatura e peso abaixo do percentil 5 para a idade, diagnóstico radiológico indicando idade óssea compatível com a idade cronológica e exames hormonais normais (TSH: 1,8 µUI/ml; T4 livre: 1,4 ng/dl; HGH: 0,1 ng/ml; IGFI: 190,9 ng/ml e IGF BP-3: 2,93 ng/L). Considerou-se baixa estatura constitucional como principal hipótese diagnóstica e pedidas avaliações das seguintes clínicas: neuropediatria, genética clínica, urologia e oftalmologia.
Avaliação Neuropediátrica: confirmou atraso de desenvolvimento, implantação baixa dos pavilhões auriculares, nariz em sela, face triangular, relação crânio-face diminuída, trofismo muscular diminuído, motricidade ativa dos seguimentos com diminuição global.
Avaliação Urológica: testículos retráteis.
Avaliação Genética: confirmada síndrome de Dubowitz.
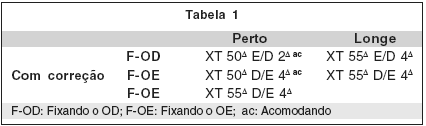
Avaliação Oftalmológica: O paciente apresentava acuidade visual (AV) para longe sem correção, feita através da tabela de Snellen, de 20/25 no olho direito (OD) e 20/100 no olho esquerdo (OE). O OE apresentava teste de "Hirschberg" com reflexo nasal de 25º. Ao exame externo: epicanto e estrabismo divergente. O "Cover test" indicava exotropia (XT) alternante com preferência pelo OD. Nada foi encontrado de anormal pela biomicroscopia e o exame de fundo de olho (FO), feito pela oftalmoscopia indireta, considerado normal em ambos os olhos (AO). A refração estática encontrada (plano no OD e -4,00 no OE) foi prescrita. O paciente foi encaminhado para teste ortóptico e posterior consulta no Ambulatório de Estrabismo da FAMEPP/UNOESTE.
O teste ortóptico revelou a AV mostrada abaixo, com informação regular pelo paciente:
Longe, com correção, angular, OD: 1,0 e OE: 0,8, e a cortical, com correção, OD: 0,7 e OE: 0,6.
Os demais exames ortópticos são demonstrados abaixo e em seqüência.
A) Avaliação motora
1 – Fixação monocular: macular em AO.
2 – Prisma e "Cover":
3 – Fixação binocular: com preferência pelo OD podendo alternar, tanto para perto quanto para longe, com ou sem correção.
4 – Ducções: normais.
5 – Versões: +2 RLD, 0/-1 OID, -2 RMD, -2 RME, 0/-1 OIE, 0/-1 RSE, +2 RLE.
6 – Medidas do desvio nas posições diagnósticas para longe com correção, não encontramos incomitâncias, nem horizontais e nem verticais, significantes.
B) Estado sensorial
Nos testes sensoriais, realizados no espaço e no sinotóforo, o paciente informou supressão alternada mais do OE, não reconhecendo visão estereoscópica.
C) Diagnóstico
Exotropia com pequeno componente vertical incomitante associado e discreta variação alfabética em A.
O paciente foi avaliado no Ambulatório de Estrabismo da FAMEPP/UNOESTE e a conduta terapêutica baseou-se em manter a prescrição da refração estática e estabelecer oclusão contínua (durante o dia todo) do OD por pelo menos 2 meses. Como a AV do menor não se alterou, manteve-se oclusão contínua do OD por mais 4 meses, retornando mensalmente ao ambulatório de estrabismo para acompanhamento.
Após 6 meses de tratamento clínico, sem mudança do ângulo de desvio, indicou-se cirurgia para correção da XT com recuo de 9 mm do músculo reto lateral esquerdo (RLE) e ressecção de 10 mm do músculo reto medial esquerdo (RME), como tentativa para correção de 50D.
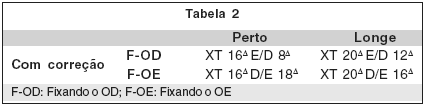
Abaixo se demonstra o resultado ortóptico do 6º mês do pós-operatório (PO), com o desvio residual do paciente.
1 - Prisma e "Cover": 6º mês de PO em OE para correção de XT.
Relata-se grande dificuldade na realização dos exames, aos quais o paciente foi submetido, devido ao quadro intenso de hiperatividade.
Apesar do ângulo ser significante, a estética do paciente está muito boa e os pais da criança, satisfeitos com o resultado cirúrgico.
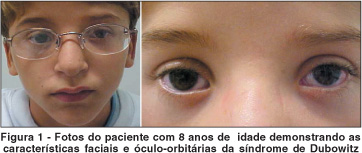
Abaixo estão representadas fotos do paciente (à esquerda, mostrando as alterações faciais características da síndrome e à direita, as conformações óculo-orbitárias).
DISCUSSÃO
O oftalmologista deve estar sempre atento às suas avaliações clínicas, já que muitas alterações oculares associadas podem indicar uma síndrome ou distúrbio genético qualquer, sendo tratada, quando ocorrer desatenção do profissional, como se fossem meras coincidências e por isso desprezadas. A não continuidade do esclarecimento sindrômico pode ser fatal se ocorrerem associações graves como: oclusão de carótida, subclávia aberrante, leucemia(10), linfoma, neuroblastoma e outras alterações que aparecem com menor freqüência na síndrome de Dubowitz. É claro que quadros clínicos crônicos de diarréia e vômitos que podem ocorrer no início da síndrome, se não tratados adequadamente podem se tornar bem mais graves do que se imagina, ainda mais em pacientes que apresentam alterações do sistema imunológico como os que possuem esta síndrome.
As manifestações oculares ocorrem em aproximadamente 20% dos pacientes com síndrome de Dubowitz e as mais comuns são: estrabismos, blefarofimoses, ptose palpebral, telecanto e prega epicântica(11). Annemans e cols. relataram uma paciente de 3 anos de idade com Dubowitz e perda da visão devido a catarata bilateral(11).
O paciente descrito neste relato apresenta alterações oftalmológicas típicas da síndrome, como estrabismo e epicanto, associadas às alterações sistêmicas que, na verdade, excluídas outras síndromes através de exames genético-clínicos, revelam o diagnóstico de síndrome de Dubowitz. As manifestações clínicas que indicam diagnóstico da síndrome neste paciente compreendem as várias alterações dos mais variados sistemas, porém de maior significância temos: peso ao nascimento de 2.200 g, tratando-se de peso pequeno para a idade gestacional (PIG), indicando retardo do crescimento pré-natal, além de ter permanecido abaixo do percentil 5 para a idade desde o nascimento, microcefalia, estatura normal ao nascimento (46 cm), mas permanecendo abaixo do percentil 5 desde 1 ano de idade, com níveis hormonais normais e padrão característico da face com forma triangular, implantação baixa dos pavilhões auriculares e base nasal achatada, além da hiperatividade associada à voz típica. Não se sabe a respeito do crescimento pré-natal do paciente já que a mãe não fez acompanhamento em nosso serviço. Sabe-se, entretanto, que o paciente nasceu com estatura normal, porém PIG, alteração não vista normalmente nesses pacientes, já que os mesmos geralmente nascem com peso dentro dos parâmetros da normalidade(7).
A exotropia foi corrigida por método cirúrgico de recuo e ressecção, porém devido às prováveis assimetrias oculares e faciais não se pôde obter resultado de excelência, até porque o paciente não fazia uso adequado, nem de óculos, quanto menos da oclusão ocular. Associado a este fato, o menor possui grande hiperatividade o que impossibilita a realização adequada dos exames oftalmológicos. É fundamental que se avaliem adequadamente esses pacientes já que as conformações ocular e orbitária são diferentes e por isso mais difícil prever resultados cirúrgicos.
Podugol´nikov e Solonichenko demonstraram a redução da concentração de heterocromatina C nos cromossomos 1, 9 e 16 em embriopatias de causas não conhecidas e em crianças com trissomia do 21. Esse fato também foi observado em crianças com retardo de crescimento associado a síndromes genéticas (síndromes de Russel-Silver, Dubowitz, etc)(12-13). O cálculo da concentração de heterocromatina C poderá ajudar na identificação dessas síndromes, melhorando assim o arsenal clínico-laboratorial em busca do diagnóstico correto.
CONCLUSÃO
Devido aos problemas oculares, os quais o paciente com esta síndrome pode vir a desenvolver ou simplesmente já possuir, achamos necessário todos os pacientes com síndrome de Dubowitz, não só serem avaliados uma única vez pelo oftalmologista, mas sim acompanhados com exames periódicos desde o nascimento. Essa avaliação deverá ser feita anualmente e de forma completa, com exame externo, refracional, biomicroscópico, de fundo de olho, ortóptico e através de exames complementares quando necessários. Salienta-se também a necessidade, mais do que provada, das avaliações multidisciplinares em casos como desta síndrome, na qual cabe, não só ao pediatra, mas sim a qualquer médico, incluindo-se o oftalmologista, o interesse em pelo menos suspeitar, ou até mesmo investigar pacientes com aspecto ou padrões anormais, tanto físico quanto psíquico, como descrito nesta síndrome.
REFERÊNCIAS
1. Dubowitz V. Familial low birthweight dwarfism with an unusual facies and a skin eruption. J Med Genet 1965;42:12-7.
2. Grosse R, Gorlin J, Opitz JM. The Dubowitz syndrome. Z Kinderheilkd 1971;110:175-87.
3. Optiz JM, Pfeiffer RA, Hermann JP, Kushnick T. Studies of malformation syndromes of man XXIV B: the Dubowitz syndrome. Further observations. Z Kinderheilkd 1973;116:1-12.
4. Majewski F, Michaelis R, Moosmann K, Bierich JR. A rare type of low birthweight dwarfism: the Dubowitz syndrome. Z Kinderheilkd 1975;120: 283-92.
5. Orrison WW, Schnitzler ER, Chun RW. The Dubowitz syndrome: further observations. Am J Med Genet 1980;7:155-70.
6. Rodden WS, Crouch ER, Leichtman LG, Becker HC. Ophthalmologic findings in the Dubowitz syndrome. J Pediatr Ophthalmol Strabismus 1999;36:37-9.
7. Tsukahara M, Opitz JM. Dubowitz syndrome: review of 141 cases 36 previously unreported patients. Am J Med Genet 1996;63:277-89.
8. Bader-Meunier B, Mielot G, Tchernia G, Lavergne J, Leonard C, Dommergues J. Dubowitz syndrome: An etiology for childhood myelodysplasia. Int J Pediatr Hematol Oncol 1996;6:105-7.
9. Kato T, Komatsu H, Sakakibara A, Tagami H. Ichthyosiform eruption in a patient with the Dubowitz syndrome. Pediatr Dermatol 1995;12:130-3.
10. Andrade-Machado R, Machado-Rojas A, de la Torre-Santos ME. Síndrome de Dubowitz, poliomiositis y leucemia mieloblástica aleucemica. Uma nueva combinacion. Rev Neurol 2001;32:500.
11. Annemans I; Foets B, Jaeken J, Casteels I. Sudden development of bilateral cataract in a child with Dubowitz syndrome: a case report. Bull Soc Belge Ophtalmol 2000;278:23-5.
12. Podugol´nikov OA, Solonichenko VG. A cytogenetic study of the functions of the variable regions in human C heterochromatin I. The effect of C heterochromatin on gene expression. Tsitologiia 1994;36:1035-40.
13. Podugol´nikov OA, Solonichenko VG. A cytogenetic study of the functions of the variable regions in human C heterochromatin III. The relationship between the amount of C heterochromatin and the occurrence of the fetal alcohol syndrome. Tsitologiia 1994;36:1049-53.
Endereço para correspondência
Vanderson Glerian Dias
Rua João Batista Colnago 190, apto. 2
Presidente Prudente (SP) CEP 19050-670
E-mail: [email protected]
Recebido para publicação em 07.11.2002
Versão revisada recebida em 24.04.2003
Aprovação em 27.08.2003
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos às Dras. Rosane da Cruz Ferreira e Ana Tereza Ramos Moreira.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.