Arq. Bras. Oftalmol. 2019; 82 (3): 10.5935/0004-2749.20190051
Total: 1969
Bahar Gunes1; Remzi Karadag2; Lokman Uzun3; Bengu Cobanoglu Sımsek4; Ayse Serap Karadag5
DOI: 10.5935/0004-2749.20190051
ABSTRACT
A 12-year-old girl presented with recurrent crusty debris and dandruff at the base of both eyelashes despite having completed different medical treatments. She had had a hoarse voice since her early childhood. Upon anterior segment examination of the eyes, we found yellow-white, bead-like papules on the margins of the eyelids. An otolaryngologist detected multiple nodules on the vocal cords and buccal mucosa. Ultrasonography revealed salivary stones in the main parotid ducts. And a dermatological examination revealed thickened skin lesions on the elbows and knees with a biopsy showing histopathological findings of lipoid proteinosis. We diagnosed the patients as having Urbach-Wiethe syndrome or lipoid proteinosis, a rare autosomal recessive multisystem disorder with variable manifestations vary that difficult the diagnosis. The ocular manifestations are not well known among ophthalmologists, but the typical lid lesions are pathognomonic and ophthalmologists should be aware of this presentation to identify patients with Urbach-Wiethe syndrome.
Keywords: Blepharitis; Eyelid; Lipoid proteinosis of Urbach-Wiethe; Case reports
RESUMO
Uma menina de 12 anos apresentava restos crostosos e caspa recorrente na base de ambos os cílios, apesar de ter completado diferentes tratamentos médicos. Ela tinha uma voz rouca desde a infância. No exame do segmento anterior dos olhos, encontramos pápulas amarelo-esbranquiçadas nas margens das pálpebras. Um otorrinolaringologista detectou múltiplos nódulos nas cordas vocais e na mucosa bucal. A ultrassonografia revelou cálculos salivares nos principais ductos parotídeos. Um exame dermatológico revelou lesões cutâneas espessas nos cotovelos e joelhos com uma biópsia mostrando os achados histopatológicos de proteinose lipoide. Diagnosticamos os pacientes da síndrome de Urbach-Wiethe ou proteinose lipoide, um distúrbio multissistêmico autossômico recessivo raro, com manifestações variáveis, que dificultam o diagnóstico. Manifestações oculares não são bem conhecidas entre oftalmologistas, mas as lesões típicas da pálpebra são patognomônicas e os oftalmologistas devem estar atentos a essa apresentação para identificar pacientes com síndrome de Urbach-Wiethe.
Descritores: Blefarite; Pálpebras; Proteinose lipoide de Urbach e Wiethe; Relatos de casos
INTRODUCTION
Urbach-Wiethe syndrome (UWS) or lipoid proteinosis is a rare autosomal recessive multisystem disorder that usually presents with dermatological, ophthalmological, otolaryngological, and neurological manifestations. It was first described by the dermatologist Urbach and the otolaryngologist Wiethe in 1929(1). Mutations in the extracellular-matrix protein-1 gene located on chromosome 1q21 are responsible for UWS(2), which is characterized by deposition of an eosinophilic hyaline-like material in various tissues of the body, but mainly on the larynx, skin, and tongue. Although the most striking symptom is hoarseness(3) that is usually noticed in infancy or early childhood, lid lesions called moniliform blepharosis are considered pathognomonic for the disease(4). We report here the case of a patient with lipoid proteinosis that presented with moniliform blepharosis and received unnecessary blepharitis treatment.
CASE REPORT
A 12-year-old girl presented with recurrent crusty debris and dandruff at the base of her eyelashes despite having had successive medical treatments such as antibiotic/steroid combinations and eyelid shampoos. She had a hoarse voice since early childhood and lacked a history of neurological problems. Upon ophthalmic examination, she had uncorrected visual acuity of 20/20 in both eyes with yellow-white, bead-like papules on the margins of the upper and lower eyelids of both eyes (Figure 1A, 1B) that we diagnosed as moniliform blepharosis. The remainder of the anterior segment examination was normal, and the fundus evaluation of both eyes was within normal limits. We requested ear, nose, and throat, dermatology and neurology consultations. The otolaryngologist found multiple nodules on the vocal cords (Figure 2A, 2B) and buccal mucosa (Figure 2C). She had swelling of the parotid glands, and ultrasonography revealed salivary stones in the main parotid ducts. A dermatological examination revealed thickened skin lesions on the elbows and knees (Figure 2D). The neurological examination was normal. A skin lesion biopsy specimen taken by a dermatologist showed histopathological findings of lipoid proteinosis with diffuse deposition of pale, eosinophilic hyaline-like substance in the superficial dermis, increased number of small capillaries, and PAS-positive diastase-resistant hyaline material around the small blood vessels (Figure 1C, 1D).
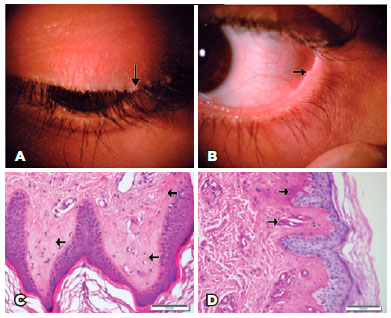
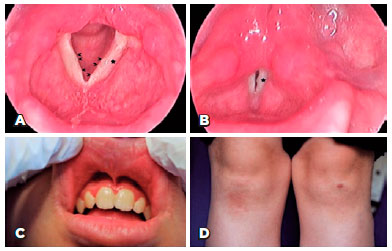
DISCUSSION
Lipoid proteinosis is a rare autosomal recessive disorder caused by a mutation in the extracellular-matrix protein-1 gene located on chromosome 1q21(2). Deposition of hyaline material composed of periodic acid-Schiff-positive glycoprotein and interspersed lipoid deposits are histopathological characteristics of the disease(5). UWS usually manifests in early childhood, but the age at diagnosis ranges from 6 months to 67 years(6). The syndrome is more common among Caucasian people(7).
The first clinical manifestation is usually hoarseness of the voice due to involvement of the upper aerodigestive tract mucosa. In our patient, the hoarseness of the voice was present since early childhood, but the patients had not received any related examinations or treatment. Skin lesions, such as skin rashes, involving the face, scalp, and trunk with a thickened, pale yellow-brown, pockmarked appearance of the lesions appear later. Our patient also had thickened skin lesions on her elbows and knees, and she had salivary stones in the main parotid gland, which caused swelling.
UWS is not well known among ophthalmologists, although its ocular manifestations are very important for the diagnosis. The most common ocular lesions include eyelid lesions, called moniliform blepharosis, which are accepted as a pathognomonic feature of the disease(4). Our patient presented moniliform blepharosis, but she had been misdiagnosed as having blepharitis by other ophthalmologists; therefore, she had been prescribed numerous useless eye drops and ointments. Less common lid lesions are infiltration of the glands of Zeiss, Moll, and Meibomian causing madarosis and trichiasis, and other ocular features, such as glaucoma and conjunctival nodules, may also be present(8). Retinitis pigmentosa, bilateral uveitis, and korektopia(9) have also been reported in the literature.
Neurological manifestations include psychiatric problems including rage attacks and paranoia, mental retardation, and temporal lobe epilepsy(10). In our case, we found none of these neurological symptoms.
In conclusion, UWS is a very rare condition, and a multidisciplinary approach is necessary for its diagnosis. As manifestations vary, the diagnosis may be difficult. Ocular manifestations are not well known among ophthalmologists, but the typical lid lesions are pathognomonic and should be better known.
REFERENCES
1. Urbach E, Wiethe C. Lipoidosis cutis et mucosae. Virchows Arch Pathol Anat. 1929;273(2):285-319.
2. Hamada T, McLean WH, Ramsay M, Ashton GH, Nanda A, Jenkins T, et al. Lipoid proteinosis maps to 1q21 and is caused by mutations in the extracellular matrix protein 1 gene (ECM1). Hum Mol Genet. 2002;11(7):833-40.
3. Di Giandomenico S, Masi R, Cassandrini D, El-Hachem M, De Vito R, Bruno C, et al. Lipoid proteinosis: case report and review of the literature. Acta Otorhinolaryngol Ital. 2006;26(3):162-7.
4. Sharma V, Kashyap S, Betharia SM, Gupta S, Pathak H. Lipoid proteinosis: a rare disorder with pathognomonic lid lesions. Clin Exp Ophthalmol. 2004;32(1):110-2.
5. Sainani MP, Muralidhar R, Parthiban K, Vijayalakshmi P. Lipoid proteinosis of Urbach and Weithe: case report and a brief review of the literature. Int Ophthalmol. 2011;31(2):141-3.
6. Irkeç M, Orhan M, Orhan D, Durgun B, Can C. Dry eye syndrome associated with Urbach-Wiethe disease. J Pediatr Ophthalmol Strabismus. 1996;33(5):265-8.
7. Rosenthal AR, Duke JR, Lipoid proteinosis: case report of direct lineal transmission. Am J Ophthalmol. 1967;64(6):1120-5.
8. Sellami D, Masmoudi A, Turki H, Mseddi M, Kammoun B, Elleuch N, et al. [Ophthalmic manifestations of lipoid proteinosis]. Presse Med. 2006;35(5 Pt 1):796-8. French.
9. Johnson LN, Hepler RS. Corectopia and lipoid proteinosis. Br J Ophthalmol.1989;73(5):394-6.
10. Di Giandomenico S, Masi R, Cassandrini D, El-Hachem M, De Vito R, Bruno C, et al. Lipoid proteinosis: case report and review of the literature. Acta Otorhinolaryngol Ital. 2006;26(3):162-7.
Submitted for publication:
August 3, 2018.
Accepted for publication:
November 15, 2018.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: None of the authors have any potential conflicts of interest to disclose.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.