Arq. Bras. Oftalmol. 2018; 81 (6): 10.5935/0004-2749.20180102
Total: 1562
Álvaro Buno Botentuit Serra de Castro1; Bruno Nobre Lins Coronado2; Ráysa Hellen Assunção Costa1; Maria Regina Chalita3; Wener Passarinho Cella1; Marcos Pereira de Avila3
DOI: 10.5935/0004-2749.20180102
ABSTRACT
Alström syndrome is a rare disorder characterized by mutations to the ALMS1 gene and clinical findings of childhood obesity, diabetes mellitus, dilated cardiomyopathy, sensorineural hearing loss, and progressive cone-rod dystrophy, which may result in blindness. Ocular manifestations occur in the first decade of life with nystagmus, blepharospasm, and photophobia leading to progressive and severe reductions in visual acuity. This study describes the retinal structure and functional aspects of four patients (8 eyes) from two different families as determined by optical coherence tomography (OCT), fundus autofluorescence, and full-field electroretinography. There was a correlation between morphological and functional findings, evidenced by typical funduscopic changes of retinal dystrophy in spectral domain-OCT and electrophysiological analyses. Foveal characteristics include a single layer of undifferentiated photoreceptors with retinal disorganization mainly from external segments, in agreement with previous reports in the literature. Fundus autofluorescence showed areas of hyperautofluorescence interspersed by hypoautofluorescence dots suggesting, respectively, involvement and atrophy of retinal pigmented epithelial cells in the macular zone. Electroretinographic analyses showed early dysfunction of the cones followed by rapid rod deterioration.
Keywords: Alström syndrome; Electrophysiology; Autofluorescence; Optical coherence tomography; Retinal dystrophy
RESUMO
A síndrome de Alström é uma doença rara caracterizada por mutações no gene AMLS 1 e achados clínicos de obesidade infantil, diabetes mellitus, cardiomiopatia dilatada, surdez neurossensorial e distrofia de cones e bastonetes progressiva, que podem resultar em cegueira. Manifestações oftalmológicas ocorrem na primeira década de vida com nistagmo, blefaroespasmo e fotofobia, levando a reduções progressivas e graves na acuidade visual. Este estudo descreve a estrutura da retina e os aspectos funcionais de quatro pacientes (oito olhos) de duas famílias distintas, conforme determinado por tomografia de coerência óptica, autoflourescência de fundo de olho e eletrorretinograma de campo total. Houve correlação entre os achados morfológicos e funcionais evidenciados por alterações fundoscópicas típicas da distrofia retiniana no domínio espectral-OCT e análises eletrofisiológicas. As características foveais incluem uma única camada de fotorreceptores indiferenciados com desorganização retiniana principalmente nos segmentos externos, de acordo com relatos prévios da literatura. A autofluorescência de fundo mostrou áreas de hiperautofluorescência, sugerindo, respectivamente, envolvimento e atrofia das células do epitélio pigmentar da retina na região macular. Análises eletrorretinográficas mostram disfunção precoce de cones, seguida de rápida deteriorização da haste.
Descritores: Síndrome de Alstrom; Eletrofisiologia; Autofluorescência; Tomografia de coerência óptica; Distrofia retiniana
INTRODUCTION
Alström syndrome (AS) is a very rare autosomal recessive disorder, first described in 1959 by Henry Alström(1,2), with an estimated prevalence of one case per 100.000 individuals. This disorder is characterized by mutations to the ALMS1 gene(3) and clinical manifestations of childhood obesity, short stature in adulthood, acanthosis nigricans, hypothyroidism(4), hepatic, pulmonary, and renal dysfunction(4,5), diabetes mellitus, dilated cardiomyopathy, scoliosis, progressive cone-rod dystrophy, which will likely result in blindness, and sensorineural hearing loss(4,6). Male hypogonadism is a variable feature(7).
Early diagnosis of AS is usually based on the phenotype and by direct sequencing analysis of the ALMS1 gene(8), but is complicated by the progressive and non-simultaneous onset of the principal symptoms. Ocular manifestations of AS occurring in the first decade of life include nystagmus, blepharospasm, and photophobia with severe reduction in visual acuity(7,9). In addition, narrowing of the retinal vessels, cone-rod retinal dystrophy, chorioretinal atrophy, atypical pigmentary retinopathy without classical bone spicules, macular cellophane-like appearance, optic atrophy revealed by a funduscopic examination, moderate or high hyperopia, and posterior subcapsular cataracts may be present(7,9,10).
This study describes the ocular findings of four patients (8 eyes) from two different families with AS, as determined by optical coherence tomography (SD-OCT), fundus autofluorescence (FAF), and full-field electroretinography (ERG).
TWO FAMILIES
Family 1: consanguinity
In family 1, AS was identified in two brothers, one girl, and one boy. The parents are cousins and no other relatives presented with AS.
Case 1
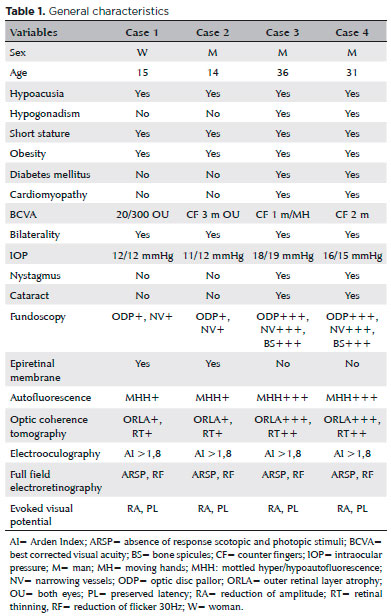
15-year-old girl with progressive vision loss since birth, photophobia, and a 1-year history of progressive bilateral hypoacusia. Other clinical manifestations of this patient included a short stature, obesity, acanthosis nigricans, and hepatic steatosis. Her best corrected visual acuity (BCVA) was 20/300 in both eyes (OU). Biomicroscopy showed no cataracts and intraocular pressure (IOP) was 12 mmHg in OU.
Case 2
14-year-old boy with progressive vision loss since birth, photophobia, and a 6-month history of bilateral hypoacusia. Additional manifestations of AS included hepatic steatosis, acanthosis nigricans, short stature, and obesity. BCVA was “counter fingers” (CF) 3 m for the right eye and CF 1 m for the left eye. Biomicroscopy showed no cataracts and IOP was 11/12 mmHg in OU.
Family 2: No consanguinity
Of three male siblings, two presented with AS. No other relatives were diagnosed with AS. There was no degree of consanguinity between the parents.
Case 3
36-year-old man with progressive vision loss since birth, nystagmus, photophobia, and progressive bilateral hypoacusia that began about 25 years ago. Other manifestations of AS included hypogonadism, short stature, obesity, diabetes mellitus, acanthosis nigricans, hepatic steatosis, and dilated cardiomyopathy. BCVA was CF 1 m for the right eye and moving hands” in left eye. Biomicroscopy showed subcapsular cataracts in OU and IOP was 18/19 mmHg.
Case 4
31-year-old man with progressive vision loss since birth, nystagmus, photophobia, and bilateral hearing loss that began about 18 years ago. Other manifestations of AS included hypogonadism, short stature, obesity, acanthosis nigricans, and hepatic steatosis. BCVA was CF 2 m in OU. Biomicroscopy showed subcapsular cataracts in OU and IOP was 16/15 mmHg.
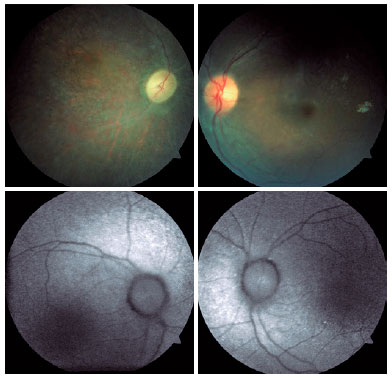
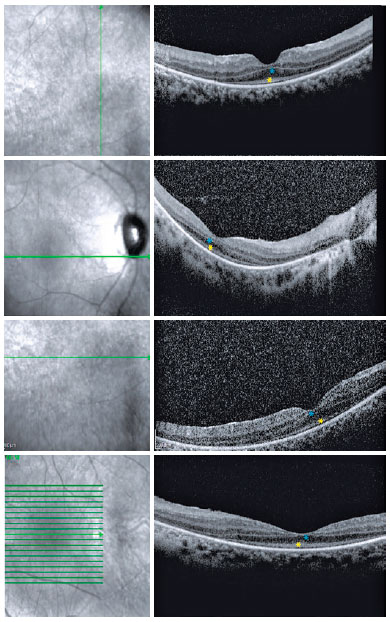
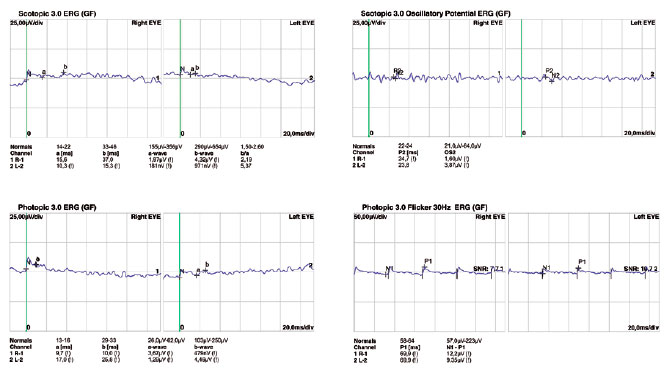
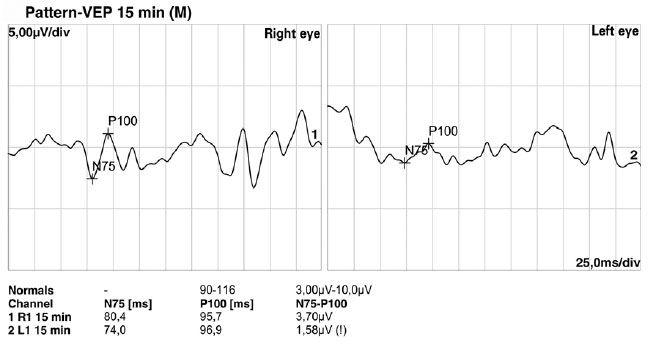
Fundoscopic findings showed optic disc pallor, narrowing of retinal vessels, and peripheral bone spicules (only in cases 3 and 4) in OU of all patients. FAF showed smooth hyper/hypoautofluorescence macular mottling (Figure 1), while SD-OCT demonstrated, in all cases, areas of outer retinal layer atrophy and central retinal thinning (Figure 2). Electrooculography demonstrated an Arden index of >1.8, besides absence of ERG registered scotopic and photopic stimuli (ARSP) responses with reduction of flicker electroretinography at 30 Hz. Reduction of amplitude and preserved latency occurred in evoked visual potential (Table 1, Figures 3 and 4).
DISCUSSION
This study describes ophthalmological findings of AS in a series of cases, which included visual acuity impairment since the first decade of life, nystagmus, photophobia, blepharospasm, optic disc pallor, narrowing of the retinal vessels, retinal pigmentary epithelium mottling, peripheral retinal bone spicules, and epiretinal membranes(7,10), besides obesity, short stature, diabetes, deafness, and absence of mental retardation. Male hypogonadism, an atypical sign(7), was present in two patients (Table 1).
There was positive correlation between morphological and functional findings. That is, retinal dystrophy and associated retinal findings, as assessed by SD-OCT, were in agreement with the electrophysiological findings. SD-OCT analyses demonstrated a single layer of undifferentiated and immature photoreceptors in the foveal zone with atrophy mainly of the peripheral retinal external layers (Figure 2). In addition, abnormal persistence of internal retinal layers occurred in the central foveal zone. Changes in FAF are poorly standardized in AS. In the presented cases, however, there was evidence of hyperautofluorescence areas and hypoautofluorescence spots suggesting, respectively, involvement and atrophy of retinal pigment epithelial cells. Electrophysiology demonstrated early dysfunction followed by rapid rod deterioration in advanced stages of disease with no response to scotopic and photopic stimuli leading a mixed dysfunction of photoreceptors(10) (Figures 1 and 3).
The main differential diagnoses of AS are Leber’s congenital amaurosis, achromatopsia, and Bardet-Biedl syndrome(7,10). With regard to Leber’s congenital amaurosis, ERG shows a pathogenic pattern in the first years of life(10). However, unlike with AS, there is no association with diabetes mellitus or deafness(7). In achromatopsia, ERG demonstrates the complete absence of cone responses and normal functioning of the rods(7), while OCT usually shows atrophy or cavitation of the external retina in the foveal zone, as well as immaturity of the inner retinal layers, but these findings are not characteristic of AS(7,10). Bardet-Biedl syndrome is an autosomal recessive disease characterized by degeneration of the retinal pigment epithelium, obesity, hypogonadism, and digital disorders. BVCA findings of Bardet-Biedl syndrome are usually a better than with AS in the first decade of life and there is a high association with mental retardation, but no correlation with diabetes or deafness(7).
The recognition of AS is hampered by its rarity and the fact that characteristic signs do not usually appear early and simultaneously, thus this disease is often retrospectively diagnosed. Early diagnosis by ophthalmologists can minimize morbidity and mortality. Therefore, AS should be a differential diagnoses in cases with dystrophy of the cones and rods in obese children with or without the presence of cardiomyopathy(10).
REFERENCES
1. Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand Suppl. 1959;129:1-35.
2. Minton JA, Owen KR, Ricketts CJ, Crabtree N, Shaikh G, Ehtisham S, et al. Syndromic obesity and diabetes: changes in body composition with age and mutation analysis of ALMS1 in 12 United Kingdom kindreds with Alström syndrome. J Clin Endocrinol Metab. 2006; 91(8):3110-6.
3. Jagger D, Collin G, Kelly J, Towers E, Nevill G, Longo-Guess C, et al. Alström Syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet. 2011;20(3):466-81.
4. Farmer A, Aymé S, de Heredia ML, Maffei P, McCafferty S, Młynarski W, et al. EURO-WABB: an EU rare diseases registry for Wolfram syndrome, Alström syndrome and Bardet-Biedl syndrome. BMC Pediatr. 2013;13(1):130.
5. Marshall JD, Paisey RB, Carey C, Macdermott C. Alstrom syndrome. In: Adam MP, Ardinger HH, Pagon RA, editors. Gene reviews(r). Seattle: University of Washington; 1993. [updated May 31, 2012].
6. Worthley MI, Zeitz CJ. Case of Alström syndrome with late presentation dilated cardiomyopathy. Intern Med J. 2001;31(9):569-70.
7. Benso C, Hadjadj E, Conrath J, Denis D. Three new cases of Alström syndrome. Graefes Arch Clin Exp Ophthalmol. 2002;240(8):622-7.
8. Kaya A, Orbak Z, Cayir A, Döneray H, Tasdemir S, Ozantürk A, et al. Combined occurrence of Alström syndrome and bronchiectasis. Pediatrics. 2014133(3):e780-3.
9. Russell-Eggitt IM, Clayton PT, Coffey R, Kriss A, Taylor DS, Taylor JF. Alström syndrome. Report of 22 cases and literature review. Ophthalmology. 1998;105(7):1274-80.
10. Marshall JD, Maffei P, Collin GB, Naggert JK. Alström syndrome: genetics and clinical overview. Curr Genomics. 2011;12(3):225-35.
Submitted for publication:
December 13, 2017.
Accepted for publication:
June 9, 2018.
Funding: No specific financial support was available for this study
Disclosure of potential conflicts of interest: None of the authors have any potential conflicts of interest to disclose

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.