Arq. Bras. Oftalmol. 2004; 67 (1): 10.1590/S0004-27492004000100029
Total: 2247
Ana Regina Cruz Vlainich1; Norma Allemann2; Isaac Neustein3
DOI: 10.1590/S0004-27492004000100029
RESUMO
Os autores descrevem associação rara bilateral de coloboma da íris, coróide, retina e cabeça do nervo óptico, microcórnea e deficiência de clivagem de câmara anterior. Fazem também uma abordagem quanto à embriologia e à dificuldade de identificar se a causa da queda contínua e insidiosa da visão é devida ao próprio coloboma ou a outra doença associada, como o glaucoma. Os exames subsidiários disponíveis como campo visual, fundoscopia e tonometria, são de pouca confiabilidade devido ao nistagmo e à baixa acuidade visual.
Descritores: Coloboma; Anormalidades múltiplas; Câmara anterior; Glaucoma; Gonioscopia; Distrofias hereditárias da córnea
ABSTRACT
The authors describe a rare association of bilateral typical coloboma, microcornea and anterior chamber cleavage deficience. They also discuss the embriology and the difficulties to identify if the continuous and insidious vision impairment is due to coloboma or to other associated disease, like glaucoma. Supplementary tests, such as visual field, ophthalmoscopy, and tonometry are not reliable because of the presence of nistagmus and low visual acuity.
Keywords: Coloboma; Abnormalities multiple; Anterior chamber; Glaucoma; Gonioscopy; Corneal dystrophies; hereditary
INTRODUÇÃO
O coloboma é uma malformação congênita que pode atingir qualquer região ocular, hereditária na forma de defeito ocular isolado, geralmente de caráter autossômico dominante com penetração incompleta ou defeito ocular associado a malformações sistêmicas ligadas a vários gens(1-2), ou adquirida pela ação de drogas e/ou agentes infecciosos, provocada pelo fechamento deficiente da fissura embrionária(2). O coloboma pode envolver um ou os dois olhos, apenas a cabeça do nervo óptico ou se prolongar anteriormente para a região ínfero-nasal, com ou sem comprometimento de íris(2-3).
As síndromes de clivagem da câmara anterior, como Axenfeld e Rieger, também são congênitas, ligadas à depleção do cromossomo 13 banda q14, ou adquiridas; e são expressões clínicas das deficiências de clivagem da câmara anterior durante a embriogênese, podendo evoluir com glaucoma, em aproximadamente metade dos casos(2,4).
RELATO DO CASO
SAV, 30 anos, sexo feminino, branca, professora, foi atendida em nosso Serviço com queixa de baixa acuidade visual desde a infância em AO, e piora há 1 ano, sem patologias sistêmicas associadas. Em seu prontuário encontrou-se acuidade visual (AV) de 20/100 A.O. há 3 anos. Ao exame clínico apresentou-se sem alterações sistêmicas. A mãe tem história de contato com rubéola durante a gravidez, mas sem exames comprobatórios. Não havia antecedentes familiares tendo sido examinados mãe, irmãos e filha, e somente a mãe havia sido facectomizada intra-capsular sem implante de lente intra-ocular aos 40 anos e apresentava à gonioscopia ligamentos pectíneos em pequena quantidade.
Ao exame ocular a paciente apresentava acuidade visual de 20/400 em ambos os olhos sem melhora com correção, com nistagmo pendular que piorava à fixação e refração -2,00 Desf em OD (olho direito) e –5,00 Desf em OE (olho esquerdo).
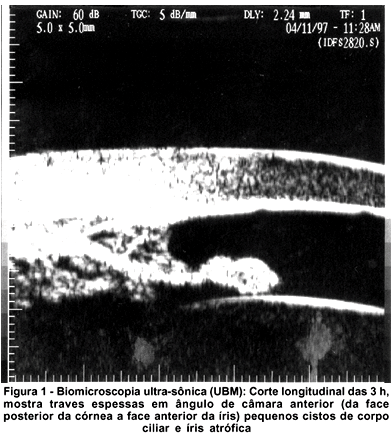
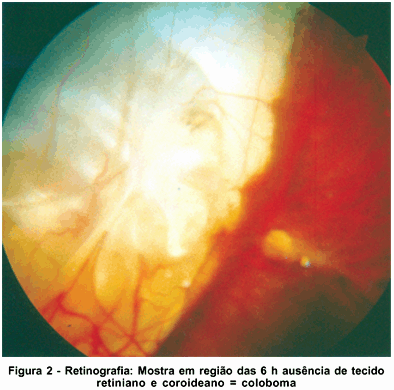
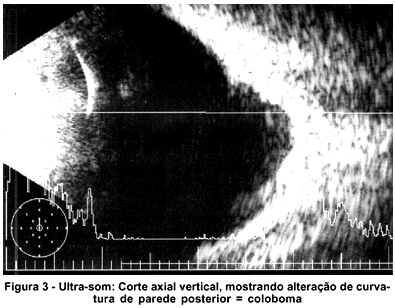
À biomicroscopia apresentou ausência dos folhetos irianos nos 2/3 centrais às 6 horas, caracterizando coloboma incompleto de íris em AO, sendo que apresentava em OD orifício inferiormente a ele, sobrando apenas pequena faixa de íris entre o coloboma de retina e íris; diâmetro corneano horizontal (branco-branco) de 8 mm, cristalino evidenciando as lamelas, sem opacidade e sem alteração da forma. À gonioscopia notou-se presença de traves espessas no ângulo, entre a íris e a face posterior da córnea, com linha de Scwalbe proeminente (embriotoxon posterior) nos 360º, confirmadas ao exame de biomicroscopia ultra-sônica de alta freqüência (UBM) onde também se evidenciaram cistos de corpo ciliar (Figura 1) e câmara anterior de profundidade diminuída (1,33 mm). À tonometria de aplanação apresentou em OD 15 e OE 16 mmHg, e em medidas posteriores níveis abaixo de 18 mmHg AO. À oftalmoscopia indireta (Figura 2) e à ultra-sonografia (Figura 3) constatou-se ausência de tecido retiniano e coroideo tipo coloboma que se estende inferiormente do nervo óptico à periferia, envolvendo mácula. Mediu-se comprimento axial de OD 22,98 mm e OE 25,49 mm (média de 5 medidas em cada olho), não caracterizando, portanto, microftalmo, mas com medidas duvidosas devido à presença do coloboma posterior. A campimetria foi de baixa confiabilidade devido ao nistagmo.
Optou-se pelo uso de betabloqueadores (Maleato de timolol 0,5% 2 vezes/dia), conforme será discutido posteriormente.
Em acompanhamento ambulatorial há 6 anos, onde todos os exames permanecem inalterados, foram prescritos óculos, não obstante a AV não melhora, pois a paciente refere conforto visual significativo.
DISCUSSÃO
A paciente apresenta coloboma, microcórnea e alteração no ângulo da câmara anterior, não sendo possível caracterizar como Síndrome de Axenfeld ou Rieger, ficando com diagnóstico generalizado de deficiência de clivagem de câmara anterior (anomalias de formação da câmara anterior). Na tentativa de elucidar a hipótese etiológica de rubéola congênita, não encontramos outras alterações como catarata, microftalmo, ou malformações sistêmicas. Não há também indicadores de hereditariedade.
Na literatura encontraram-se, apenas, duas descrições semelhantes em seres humanos; ambas apresentavam coloboma apenas de íris, sendo que uma delas apresentava também outras múltiplas malformações sistêmicas e se relacionava com a depleção do cromossomo 6p(5-6). Em estudo genético experimental do gem Pitx2, um dos ratos apresentou, associado à Síndrome de Rieger, coloboma de nervo óptico e ausência de músculos extra-oculares, sendo que o próprio autor descreve estas alterações como inesperadas(7).
Na tentativa de explicar embriologicamente tal associação entende-se que a fissura embrionária deveria se fechar da parte central da vesícula óptica em direção anterior até a íris e posterior até o nervo óptico. Na formação do coloboma não há fechamento, faltando o ectoderma superficial e, consequentemente, o epitélio pigmentar da retina não se formará. O mesoderma penetraria na parte posterior da fissura originando a coróide, e na parte anterior para formar o estroma iriano, corneano e, com a separação, o ângulo da câmara anterior (esses processos ocorrem entre a quarta e sexta semana embrionária). No coloboma, a coróide também irá faltar, pois, aparentemente para a formação da coróide é preciso haver contato do mesoderma com epitélio pigmentar(2). Como explicação para as alterações de clivagem da câmara anterior, alguns autores têm sugerido que o tecido resquicial no ângulo seria de estruturas anteriores derivadas da crista neural e aconteceria tardiamente na gestação(4).
Poderia, então, a não formação da coróide do mesênquima posterior ter influenciado a deficiência na formação das estruturas do mesênquima anterior ou seriam apenas anomalias coincidentes?
Quanto à clínica da paciente, o diagnóstico de glaucoma não pôde ser confirmado, pois a papiloscopia não forneceu base comparativa; a tonometria estava dentro dos padrões e o campo visual não foi confiável, devido ao nistagmo, baixa acuidade e alterações próprias do coloboma. Como existe alta incidência de glaucoma em pacientes com Síndromes de Axenfeld e Rieger(2,4-5) decidiu-se pelo tratamento com beta- bloqueadores.
A associação entre coloboma e alterações de clivagem de câmara anterior é raramente encontrada na literatura(5-7). Sabendo que os colobomas podem cursar com baixa gradativa da acuidade visual, e que as alterações de clivagem podem levar ao desenvolvimento de glaucoma, fica difícil identificar se a causa da queda visual é por um desses fatores ou ambos. Tendo sempre em mente que a diminuição da visão já comprometida é muitas vezes mal diagnosticada, é preferível o tratamento do glaucoma como no caso descrito, mesmo sem certeza diagnóstica, a negligenciar, e perder visão.
REFERÊNCIAS
1. Tandon RK, Burke JP, Strachan IM. Clefting syndrome with typical and atypical irido-retinochoroidal colobomatous defects. J Pediatr Ophthalmol Strabismus 1994;31:120-2.
2. Dryja TP. Aproaches to the identification of genes responsable for hereditary ophthalmology disease. In: Albert DM, Jakobiec FA. Principals and practice of Ophthalmology. Basic science. (CD-ROM). 2nd ed. Boston: WB Saunders; 2000.
3. Alves ACS, Sallum JF, Santos BW, Erwenne CM. Associação de malformações oculares congênitas: coloboma, microftalmo, e microcórnea- relato de cinco casos. Arq Bras Oftalmol 1995;58:425-8.
4. Shields MB. Axenfeld-Rieger syndrome. A theory of mechanisms and distinction from iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc 1983;81:736-84.
5. Ozeki H, Shirai S, Ikeda K, Ogura Y. Anomalies associated with Axenfeld-Rieger syndrome. Graefs Arch Clin Exp Ophtalmol 1999;237:730-4.
6. Davies AF, Mirza G, Sekhon G, Turnpenny P, Leroy F, Speleman F, et al. Delineation of two distintic 6p deletion syndromes. Hum Genet 1999;104:64-72.
7. Gage PJ, Suh H, Camper SA. Dosage requirement of Pitx2 for development of multiple organs. Development 1999;126:4643-51.
Endereço para correspondência
Ana Regina Cruz Vlainich
Departamento de Oftalmologia - Ambulatório
Av. Ibirapuera, 981
São Paulo (SP) - CEP 04029-000
Email: [email protected]
Recebido para publicação em 28.11.2002
Versão revisada recebida em 08.05.2003
Aprovação em 28.06.2003
Trabalho realizado no Hospital do Servidor Público Estadual de São Paulo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.