Arq. Bras. Oftalmol. 2003; 66 (4): 10.1590/S0004-27492003000400009
Total: 2364
Nichard Unonius1; Michel Eid Farah2; Juliana M. Ferraz Sallum1
DOI: 10.1590/S0004-27492003000400009
RESUMO
OBJETIVO:Organizar um banco de dados regional de todos os indivíduos portadores de doenças degenerativas da retina, com o objetivo de classificar cada paciente de acordo com o tipo de distrofia e padrão de herança. MÉTODOS: Durante o encontro do Grupo Retina São Paulo no dia 5 de maio de 2001, duzentas e quarenta e três pessoas foram registradas, sendo que parte forneceu dados de antecedentes oculares, pessoais e familiares e árvore genealógica. Noventa e três pacientes foram questionados quanto a idade, origem, tipo de distrofia, história familiar e árvore genealógica, tipo de herança, outras anomalias sistêmicas e exames complementares. Foram classificados quanto ao diagnóstico e padrão de herança. RESULTADOS: Dos duzentos e quarenta e três pacientes registrados, as distrofias encontradas foram retinose pigmentária, doença de Stargardt, síndrome de Usher, amaurose congênita de Leber e coroideremia. Quanto à divisão por doença dos 93 pacientes argüidos, havia 62 pacientes com retinose pigmentária, 13 com doença de Stargardt, 13 com síndrome de Usher, três com amaurose congênita de Leber e dois com coroideremia. Dos pacientes com retinose pigmentária, o padrão de herança detectado foi autossômico dominante em quatro casos (7%), autossômico recessivo em vinte casos (32%), ligado ao cromossomo X recessivo em sete casos (11%), caso isolado em vinte e nove (47%) e padrão indeterminado em dois (3%). Para a doença de Stargardt três indivíduos (23%) seguiam o padrão de herança autossômico recessivo e dez (77%) eram casos isolados. Dos treze pacientes com síndrome de Usher, oito (61,5%) apresentavam herança autossômica recessiva, quatro (31%) eram casos isolados e um (7,5%) tinha o padrão de herança indeterminado. Os dois pacientes com coroideremia seguiam o padrão de herança ligado ao X recessivo. Para amaurose congênita de Leber, um paciente (33,5%) tinha padrão autossômico recessivo de herança e dois (66,5%) eram casos isolados. CONCLUSÃO: Destaca-se assim a importância desta classificação como a primeira referência nacional dos padrões de hereditariedade das distrofias retinianas do país. Este é o primeiro passo para se proceder em seguida a classificação genético-molecular baseada no seqüenciamento de cada gene responsável por cada um dos padrões de herança. A freqüência de cada tipo específico é semelhante à encontrada em outros trabalhos epidemiológicos de outros países.
Descritores: Doenças retinianas; Degeneração retiniana; Degeneração retiniana; Retinite pigmentosa; Retinite pigmentosa; Coroideremia; Cegueira
ABSTRACT
PURPOSE: To organize a regional data bank of all individuals that have retinal degenerative diseases, with the aim to classify each patient according to the type of distrophy and pattern of inheritance. METHODS: During the meeting of the São Paulo Retina Group on May 5th, 2001, two hundred and forty-three persons were registered, part of whom provided information concerning ocular, personal and family history and family tree. Ninety-three patients were asked about age, origin, type of dystrophy, family history and family tree information, type of inheritance, other systemic abnormalities and complementary examination. They were classified according to the diagnosis and pattern of inhe-ritance. RESULTS: The distrophies found in the registered two hundred and forty-three patients, were: retinitis pigmentosa, Stargardt disease, Usher syndrome, Leber congenital amaurosis and choroideremia. Of the ninety-three patients examined on the same day, sixty-two had retinitis pigmentosa, thirteen had Stargardt disease, thirteen had Usher syndrome, three had Leber congenital amaurosis and two had choroideremia. The inheritance pattern of the patients with retinitis pigmentosa was autosomal dominant in 4 cases (7%), autosomal recessive in twenty cases (32%), X-linked recessive in 7 cases (11%). Twenty-nine cases were isolated (47%) and two had an indeterminate pattern of inheritance (3%). Of the Stargardt disease patients, three (23%) were autosomal recessive and ten (77%) were isolated cases. Of the thirteen patients with Usher syndrome, eight (61.5%) were autosomal recessive, four (31%) were isolated cases and one (7.5%) did not have a determined inheritance pattern. The two patients with choroideremia were X-linked recessive. In Leber congenital amaurosis one (33.5%) was autosomal recessive and two (66.5%) were isolated cases. CONCLUSION: This study highlights the importance of this classification as being the first reference of inheritance patterns of retinal distrophies in our country. This is the first step to further classify the genetic and molecular characteristics based on the sequencing of each gene that causes each inheritance pattern. The frequency of each disease is similar to that of the literature.
Keywords: Retinal diseases; Retinal degeneration; Retinal degeneration; Retinitis pigmentosa; Retinitis pigmentosa; Choroideremia; Blindness
INTRODUÇÃO
Avanços recentes em genética molecular tornaram possível identificar genes responsáveis por diversas afecções humanas, dentre elas retinose pigmentária, doença de Stargardt, distrofia macular viteliforme de Best e retinoblastoma(1).
Dentre outros foram identificados genes causadores de distrofias da retina como, por exemplo, alguns tipos de retinose pigmentária, síndrome de Usher(2), doença de Stargardt, doença de Best, síndrome de Bardet-Biedl/Laurence Moon e até recentemente degeneração macular relacionada à idade(3). Esses avanços estabeleceram clinicamente o conceito de que a retinose pigmentária não é uma doença, mas o nome dado a uma série de desordens que cursam com perda de visão devido à degeneração dos fotorreceptores e epitélio pigmentado da retina.
Tem sido então necessário classificar os diferentes tipos(4) e suas síndromes relacionadas. Somente assim a base genética pode ser relacionada aos achados bioquímicos, de biologia molecular e de fisiologia. Já foi proposta classificação da retinose pigmentária baseada nos sistemas funcionais afetados pelas proteínas mutadas(4). Tal classificação conecta diretamente os vários fenótipos às mutações observadas.
Portanto classificar as distrofias a partir da pesquisa dos genes ou genotipagem é fundamental para se chegar aos tratamentos. Um exemplo é a própria retinose pigmentária, que se subdivide em vários tipos, causados por mutações genéticas em diferentes cromossomos, as quais dão à doença características próprias (desenvolvimento da doença na infância ou juventude, progressão lenta ou rápida, comprometimento da visão central e periférica).
O entendimento dos mecanismos que levam às distrofias avançou muito nos últimos 15 anos devido às técnicas da biologia molecular(1).
São muitos os temas apresentados atualmente sobre terapia genética e biologia molecular, prótese visual, experimentos com transplante de células da retina, testes com medicamentos e substâncias nutrientes para compor a dieta dos portadores. Formam-se assim quatro linhas de pesquisa em torno do assunto.
Existe agora o momento de transição das pesquisas de laboratório para os experimentos clínicos, visando encontrar os tratamentos para deter a progressão e curar essas doenças. Bem claro é o vínculo que deverá existir entre o trabalho do geneticista e do oftalmologista na descoberta do tipo de doença. Ao geneticista caberá detectar o gene causador da doença. Ao oftalmologista caberá a tarefa de traçar o quadro clínico do doente, verificar os exames e classificar o fenótipo da doença, de posse do genótipo do paciente. São numerosos os tipos de retinose, sendo que para cada fenótipo poderá haver um medicamento ou forma de abordagem (clínica, cirúrgica, terapia genética), ou talvez um mesmo tipo de abordagem para todos eles.
Fundações internacionais como a Foundation Fighting Blindness já destinaram milhões de dólares às pesquisas. Entidades como a Retina International - International Retinitis Pigmentosa Association - e em nível nacional o Grupo Retina Rio, por exemplo, procuram organizar informações, agrupar pacientes, visando a obtenção de recursos e de estrutura para a pesquisa na área das distrofias da retina. A organização permite mantê-los com informações atualizadas em relação às pesquisas e proporcionar treinamento em visão subnormal. Possíveis vantagens para os integrantes e para a sociedade científica são baseadas na importância em se conhecer o diagnóstico correto de cada um, já que o tratamento pode advir do conhecimento da fisiopatogenia a partir do erro genético; o que não quer dizer que o tratamento será diferente para cada erro genético. Pode haver um mecanismo fisiopatogênico comum a mais de um erro, agrupando pacientes numa síndrome, que possa ser modulado por uma droga que possa ser usada como tratamento.
Ao oftalmologista cabe a importante função de identificar e classificar os quadros oculares, visando junto com o geneticista a informação precisa da característica fenotípica e genotípica individual, visando a terapia específica. Porém, classificações baseadas somente na clínica podem nem sempre ter a correlação direta com o defeito genético(1).
O grupo Retina Brasil, uma entidade nacional de portadores de doenças degenerativas da retina, tem como um de seus objetivos organizar um banco de dados nacional de todos os indivíduos com doenças degenerativas da retina no Brasil, visando classificar cada paciente quanto ao tipo de distrofia e padrão de herança.
O objetivo deste estudo é apresentar os dados do primeiro levantamento realizado nos grupos regionais Retina São Paulo e Retina Vale do Paraíba.
MÉTODOS
Durante evento do Grupo Retina São Paulo em 5 de maio de 2001, realizou-se o cadastro de duzentos e quarenta e três indivíduos, dos quais parte se prestou, no dia, a fornecer dados para realização de seu histórico pessoal, familiar, heredograma. Tomaram-se no dia os dados de 93 pacientes referentes a sexo, idade, procedência, tipo de distrofia, histórico familiar e heredograma, tipo de herança, outros sintomas ou alterações sistêmicas associadas e exames complementares. O tipo de herança foi estabelecido pelos autores após análise genética dos heredogramas(5).
RESULTADOS
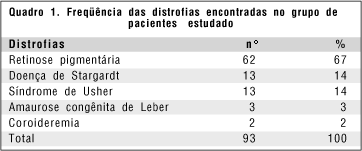
Quanto ao tipo de distrofia do total de 243 pacientes cadastrados, as apontadas foram retinose pigmentária, doença de Stargardt, síndrome de Usher, amaurose congênita de Leber e coroideremia.
Quanto à divisão por doença dos 93 pacientes argüidos, havia 62 pacientes com retinose pigmentária, 13 com doença de Stargardt, 13 com síndrome de Usher, três com amaurose congênita de Leber e dois com coroideremia (quadro 1).
Dos pacientes com retinose pigmentária (RP), o padrão de herança detectado foi autossômico dominante em quatro casos, autossômico recessivo em vinte casos, ligado ao cromossomo X recessivo em sete casos, caso isolado em vinte e nove e padrão indeterminado em dois (quadro 2). Para a doença de Stargardt três indivíduos seguiam o padrão de herança autossômico recessivo e dez eram casos isolados. Dos treze pacientes com síndrome de Usher, oito apresentavam herança autossômica recessiva, quatro eram casos isolados e um tinha o padrão de herança indeterminado. Os dois pacientes com coroideremia seguiam o padrão de herança ligado ao X recessivo. Para amaurose congênita de Leber, um paciente tinha padrão autossômico recessivo de herança e dois eram casos isolados (quadro 2).
DISCUSSÃO
A classificação fenotípica dos pacientes brasileiros portadores de distrofias é de importância capital para sua futura genotipagem. Vários centros mundiais estão coletando sangue dos pacientes para a pesquisa genética.
O atendimento destes pacientes poderá ser desenvolvido com centros aptos ao diagnóstico por meio de exames subsidiários como retinografia, angiofluoresceinografia, exames eletrofisiológicos, campo visual, teste de cores, exame genético clínico com geneticista.
O sistema de telemedicina quando desenvolvido ajudará na orientação destes pacientes e seus médicos.
Entre nós, a doença relatada com maior freqüência foi a retinose pigmentária que é o nome dado a uma série de doenças que têm em comum degeneração retiniana progressiva caracterizada por perda progressiva do campo visual, cegueira noturna e retinopatia com deposição de pigmento na retina. Esta ampla definição engloba grande número de formas oculares isoladas e secundárias que são parte de uma síndrome ou doença sistêmica(6-9), como por exemplo, amaurose congênita de Leber, doença de Goldmann-Favre, síndrome de Usher e síndrome de Bardet-Biedel. A maior parte se manifesta na fase adulta, mas menos comumente pode ser congênita ou manifestar-se na infância com perda progressiva da função dos fotorreceptores e células do epitélio pigmentário da retina. VanTrigt (1853) e Ruete (1854) após a descoberta do oftalmoscópio direto, foram os primeiros a descrever quadros de degeneração pigmentária do fundo de olho. Porém foi Donders (1855-1857) que os denominou retinitis pigmentosa. O termo retinite é inadequado, pois passa a idéia de que a inflamação é a causa dessa desordem, considerada uma distrofia ou degeneração determinada geneticamente(6). Ao passo que a designação retinose pigmentária transmite a idéia de degeneração associada a alterações da pigmentação.
Geralmente difusa, bilateral, simétrica com acometimento principal dos bastonetes. A tríade clássica é estreitamento vascular, pigmentação tipo espículas ósseas, palidez de disco. Aos 30 anos 75% dos pacientes são sintomáticos. O envolvimento se inicia do equador seguindo anterior e posteriormente, até que uma pequena ilha central de visão permaneça. Os 3 tipos de maculopatia que podem aparecer são a atrófica, membrana epiretiniana e com edema cistóide da mácula. Em geral o eletrorretinograma escotópico é inicialmente alterado, enquanto o fotópico é alterado nas fases mais tardias da doença. É encontrada em todas as raças, sem predomínio em nenhuma(10). Na forma dominante os sintomas têm início mais precocemente e o quadro é mais dramático quanto à acuidade visual final. Pacientes com retinose pigmentária perdem em média 4,6% do campo de visão residual ao ano.
O padrão de herança da retinose pigmentária pode ser caso isolado, autossômico dominante, autossômico recessivo, ligado ao X recessivo(11). Quanto aos genes responsáveis pela retinose pigmentária, uma grande quantidade já foi identificada(4) (quadro 3).
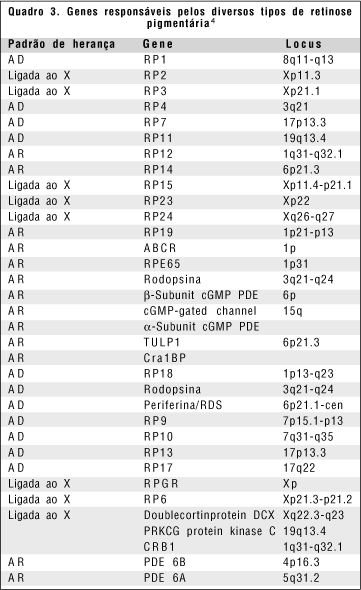
Subdivisões genéticas dos tipos de retinose pigmentária foram até então baseadas na forma de segregação dentro da família (autossômica dominante, recessiva ou ligada ao X); nenhum tipo de divisão clara de acordo com a relação genótipo-fenótipo foi feita até agora. Mesmo porque, dos 23 locus distintos reconhecidos, apenas para 12 locus os genes causadores e suas mutações foram identificados.
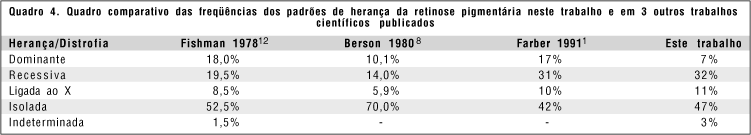
O padrão de herança mendeliana mais comum nos EUA(1) é o autossômico recessivo com 31%. Autossômico dominante representa 17% e recessivo ligado ao X representa 10%. Muitos pacientes (42%) não têm histórico familiar e são casos isolados(12).
Houve correlação entre as porcentagens de herança aqui descritas e as da literatura (quadro 4).
A doença de Stargardt, originalmente descrita por Stargardt em 1909, é juntamente com a degeneração macular relacionada à idade a distrofia macular hereditária mais comum e representa 7% das distrofias de retina. Tipicamente os pacientes apresentam baixa de visão central, usualmente na primeira ou segunda décadas de vida. Fundoscopicamente há alterações pigmentares da fóvea, com atrofia da área macular, onde uma lesão ovalada na área macular de cerca de 1,5 DP se desenvolve, com reflexo de cobre batido, que pode ou não ser circundado por "flecks" que são lesões branco-amareladas subretinianas, com formato em escamas de peixe. A acuidade visual nas fases avançadas varia entre 20/200 e 20/400. A angiografia fluoresceínica demonstra fenômeno de silêncio coroidal. A existência de "flecks" isolados sem a lesão macular em aspecto de cobre batido foi descrita depois como fundus flavimaculatus por Franceschetti em 1963. Hoje em dia é geralmente aceito que as duas entidades são diferentes apresentações de uma mesma entidade, que podem aparecer simultaneamente em membros de uma mesma família. Mesmo genes de formas dominante(13) e recessiva (como STGD3 e STGD1 ou ABCR, ver adiante) podem coexistir num mesmo indivíduo e interagir de forma a intensificar a gravidade das manifestações. A maior parte dos casos têm se apresentado como autossômica recessiva. Os achados deste trabalho são comparáveis aos da literatura, sendo que aqui o modelo de herança encontrado, quando possível de ser determinado, foi o modelo autossômico recessivo. Recentemente, um gene foi isolado no braço curto do cromossomo 1 (1p - o gene STGD1, ou ABCR) para estes casos. Recentemente o gene ABCR foi clonado, sendo que ele codifica uma proteína transmembrânica expressa exclusivamente nos bastonetes da retina e pertence à superfamília ATP-binding cassete (ABC) transporter. Um alelo do gene ABCR foi demonstrado carregando uma mutação (Arg152X, onde Arg corresponde ao aminoácido arginina, 152 corresponde à localização na proteína e X representa aminoácido mutado indeterminado) em membros de uma família, sendo que o avô de um dos pacientes com esta mutação desenvolveu degeneração macular relacionada à idade (DMRI), consistente com observações anteriores de que algumas variantes no gene ABCR podem aumentar a susceptibilidade para DMRI no estado de heretozigose(14). Já nas formas dominantes, diversos genes já foram implicados. Dois genes foram localizados, no braço longo do cromossomo 6 (6q – STGD3) e no braço longo do cromossomo 13 (13q – STGD2)(14).
No fundus flavimaculatus15 a apresentação pode ser mais tardia, durante a quarta e quinta décadas. O prognóstico costuma ser bom, com acuidade visual freqüentemente igual ou melhor do que 20/40.
A síndrome de Usher é um grupo de desordens autossômicas recessivas caracterizadas por perda congênita neurossensorial da audição, disfunção vestibular, associada à retinose pigmentária. Von Graefe foi o primeiro a reconhecer simultaneamente a associação em 1858, e em 1935 Usher descreveu a natureza hereditária desta síndrome, que levou seu nome. Sua prevalência é de cerca de 1/100000. Corresponde a 5% do total de crianças com surdez profunda, sendo responsável por cerca da metade dos casos de cegueira associada a surdez. A tipo 1 (USH1) caracteriza-se por surdez congênita profunda, arreflexia vestibular e manifestação pré-puberal de retinose pigmentária progressiva. A tipo 2 (USH2) apresenta surdez congênita moderada a severa, respostas vestibulares normais e retinose pigmentária progressiva na adolescência. A tipo 3 (USH3) se caracteriza por perda auditiva progressiva, respostas vestibulares variadas e retinose pigmentar na adolescência(2). Os seis locus para a USH1 (dos genes USH1 A até F), são localizados respectivamente nos cromossomos 14q32, 11q13, 11p15, 10q, 21q21 e 10p. Os locus para a USH2 (genes USH2A, USH2B e USH2C) foram mapeados nos cromossomos 1q41 e 5q11-13. O gene da USH3 está locado no cromossomo 3q25. O primeiro a ser identificado foi o gene USH1B localizado no cromossomo 11q13.5, ocupando mais de 100 kb de DNA genômico. Quarenta e oito dos 49 exons contêm seqüências codificadoras. O gene em questão foi denominado MYO7A(2). Ele pertence a uma classe chamada superfamília da miosina, sendo que esta última interage com a actina. A proteína do MYO7A possui partes, sendo a primeira chamada motor, que contém 729 aminoácidos. Adjacente à primeira parte há a segunda, de 129 aminoácidos. A cauda contém 1360 aminoácidos. Já foram descritas mais de 90 mutações no gene MYO7A.
Dentro da casuística apresentada neste trabalho 61,5% dos pacientes tinham herança autossômica recessiva, havia 31% de casos isolados e 7,5% com padrão indeterminado de herança.
A amaurose congênita de Leber (retinose pigmentária congênita), clinicamente, é um grupo heterogêneo de degenerações retinianas que se manifesta precocemente e de forma extremamente severa. Sua forma típica consiste em um neonato sem visão e com ERG extinto. O fundo de olho pode parecer normal no início, mas depósitos pigmentares em geral aparecem. Nistagmo se desenvolve até os 2 anos de vida. Uma segunda forma seria a de criança com cegueira noturna, com perda de campo visual a ser notada ao redor dos 4 anos de idade. Na evolução haverá quadro de adulto jovem com presença de campo tubular e visão entre 20/70 e 20/200. Outra forma consiste em degeneração pigmentária e coloboma macular, podendo cursar com ceratocone e alterações esqueléticas. Há uma forma rara em que os pacientes permanecem com a retina de aspecto normal até a idade adulta, com contração campimétrica progressiva até evoluir para cegueira noturna. A maior parte dos casos é autossômica recessiva. Na casuística apresentada um paciente tinha herança recessiva e dois eram casos isolados. O primeiro gene foi mapeado no cromossomo 17p em 1995, depois denominado GUCY2D (ou também RetGC1), que codifica a guanilato ciclase fotorreceptor-específica envolvida na fototransdução. Outros genes são o RPE65, CRX, TULP1, AIPL1 e o agora descrito CRB1(16). Ultimamente, o objetivo principal em se definirem as mutações genéticas é o de possibilitar o surgimento de meios para estratégias terapêuticas. Daí a importância de serem conhecidos e comparados a função e produtos de cada gene. O produto do gene GUCY2D já foi mencionado. O gene RPE65 é específico para o epitélio pigmentar da retina e pensa-se catalisar passo crítico no metabolismo da vitamina A, enquanto o CRX é um fator de transcripção que regula a expressão de certas proteínas dos segmentos externos dos fotorreceptores. A função dos genes CRB1, TULP1 e AIPL1 na retina ainda não foi bem estabelecida, mas acredita-se haver envolvimento no desenvolvimento ou manutenção do sistema de fototransdução.
CONCLUSÃO
Destaca-se assim a importância desta classificação como a primeira referência nacional dos padrões de hereditariedade das distrofias retinianas do país. Este é o primeiro passo para se proceder em seguida a classificação genético-molecular baseada no sequenciamento de cada gene responsável por cada um dos padrões de herança.
A freqüência de cada tipo específico é semelhante à encontrada em outros trabalhos epidemiológicos de outros países.
REFERÊNCIAS
1. Farber DB, Heckenlively JR, Sparkes RS, Bateman JB. Molecular genetics of retinitis pigmentosa. West J Med 1991;155:388-99
2. Eudy JD, Sumegi J. Molecular genetics of Usher syndrome. Cell Mol Life Sci 1999;56:258-67.
3. Zhang K, Nguyen TH, Crandall A, Donoso LA. Genetic and molecular studies of macular dystrophies: recent developments. Surv Ophthalmol. 1995; 40:51-61.
4. van Soest S, Westerveld A, de Jong PT, Bleeker-Wagemakers EM, Bergen AA. Retinitis pigmentosa: defined from a molecular point of view. Surv Ophthalmol 1999;43:321-34.
5. Beiguelman B. O registro gráfico da história genética do paciente. In: Beiguelman B. Genética médica: dinâmica dos genes nas famílias e nas populações. São Paulo: Edart; 1977. v.2, p. 113-37.
6. Pagon RA. Retinitis pigmentosa. Surv Ophthalmol 1988;33:137-77.
7. Heckenlively JR, Yoser SL, Friedman LH, Oversier JJ. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol 1988;105: 504-11.
8. Berson EL, Rosner B, Simonoff E. Risk factors for genetic typing and detection in retinitis pigmentosa. Am J Ophthalmol 1980;89:763-75.
9. Dryja TP, Berson EL. Retinitis pigmentosa and allied diseases. Implications of genetic heterogeneity. Invest Ophthalmol Vis Sci 1995;36:1197-20.
10. Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 1984;97:357-65.
11. Fishman GA, Weinberg AB, McMahon TT. X-linked recessive retinitis pigmentosa. Arch Ophthalmol 1986;104:1329-35.
12. Fishman GA. Retinitis pigmentosa – Genetic percentages. Arch Ophthalmol 1978;96:822-6.
13. Cibis GW, Morey M, Harris DJ. Dominantly inherited macular dystrophy with flecks (Stargardt). Arch Ophthalmol 1980;98:1785-9.
14. Zhang K, Kniazeva M, Hutchinson A, Han M, Dean M, Allikmets R. The ABCR gene in recessive and dominant Stargardt diseases: a genetic pathway in macular degeneration. Genomics 1999;60:234-7.
15. Fishman GA. Fundus flavimaculatus. A clinical classification. Arch Ophthalmol 1976;94:2061-7.
16. Gamm DM, Thliveris AT. Implications of genetic analysis in Leber congenital amaurosis. Arch Ophthalmol 2001;119:426-7.
17. Jay M, Evans K. Retinal dystrophies and genetic counselling. Acta Ophthalmol Scand 1996; (Suppl 219):5-7.
Endereço para correspondência
Av. Indianópolis, 1797
São Paulo (SP) - CEP 04063-003
E-mail: [email protected]
Recebido para publicação em 16.04.2002
Aceito em 30.10.2002
Trabalho realizado no Setor de Retina e Vítreo da Universidade Federal de São Paulo - UNIFESP.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos à Dra. Maria Teresa B. C. Bonanomi.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}