Arq. Bras. Oftalmol. 2002; 65 (6): 10.1590/S0004-27492002000600014
Total: 1992
Dario Grechi Goulart1; Cleide Myai1; Denise Atique4; Walter Yukihiko Takahashi2; Teruo Aihara1
DOI: 10.1590/S0004-27492002000600014
RESUMO
Relatamos o caso de uma paciente com sintomas de cegueira noturna congênita estacionária (CNCE) no qual constatamos alterações oftalmoscópicas peculiares da doença de Oguchi, inédita, nos limites de nosso conhecimento, à consideração da literatura médica brasileira publicada. Observamos e documentamos as alterações clássicas desta doença e do seu importante comemorativo, o também infreqüente, fenômeno de Mizuo-Nakamura. O exame eletrorretinográfico (ERG) também mostrou sinais típicos. Acrescentamos, brevemente, revisão bibliográfica da doença.
Descritores: Cegueira noturna; Adaptação à escuridão; Eletrorretinografia; Relato de caso; Adulto; Feminino
ABSTRACT
We herein describe a case of a patient whose only symptom was nyctalopia. Peculiar ophthalmoscopic findings of Oguchi's disease were observed. Classic changes in the electroretinogram (ERG) and Mizuo-Nakamura's phenomenon were also reported. To the best of our knowledge, there isn't another occurrence in our published literature. A brief bibliographic review is added.
Keywords: Night blindness; Dark adaptation; Electroretinography; Case report; Adult; Female
INTRODUÇÃO
Aa doença de Oguchi é caracterizada por cegueira noturna não-progressiva ou estacionária (CNCE), por descoloração acinzentada, mosqueada da retina e pelo fenômeno de Mizuo-Nakamura(1-2). Alterações típicas do eletrorretinograma também são encontradas(3-5). Relatamos, a seguir, o caso de uma paciente com queixa subjetiva de cegueira noturna e achados típicos da doença de Oguchi, condição sabidamente rara no hemisfério ocidental e, nos limites do nosso conhecimento, inédita em nossa bibliografia, o que foi a principal motivação de publicar este relato.
RELATO DO CASO
E.M.A.R., sexo feminino, branca, 38 anos, deu entrada no serviço de oftalmologia do Hospital da Santa Casa de São Paulo em julho de 2001 com queixa de baixa acuidade visual, que piora acentuadamente à noite, desde criança. Negou doenças sistêmicas ou outros antecedentes pessoais. Refere pais e avós não consangüíneos, sem doenças oculares ou sistêmicas, embora tenha um dos irmãos com problemas na visão semelhantes aos dela.
Ao exame externo, mostrou AV em AO (S/C): 20/30 e AO (C/C): +0,75E » 0,75C (180º) (20/20). A motilidade ocular extrínseca, reflexos fotomotores direto e consensual e biomicroscopia do segmento anterior apresentaram-se dentro dos limites da normalidade. Apresentou PIO de 14 mmHg AO (9 h).
Ao exame fundoscópico em AO, apresentou disco óptico normal em tamanho e coloração, com bordas nítidas e escavação fisiológica de 0,3. O tapete retiniano mostrou coloração amarelo-metálica mosqueada, de um dourado esvanecente que poupava o pólo posterior e se acentuava na médio-periferia, principalmente sobre as arcadas vasculares temporais, as quais contrastavam intensamente com o restante da retina, aparentando coloração mais escurecida que o normal. No OD foi constatada a presença de algumas drusas adjacentes à arcada vascular temporal superior. A tortuosidade vascular e o brilho macular mostraram-se preservados, como pode ser observado nas retinografias tomadas (Figuras 1 e 2).
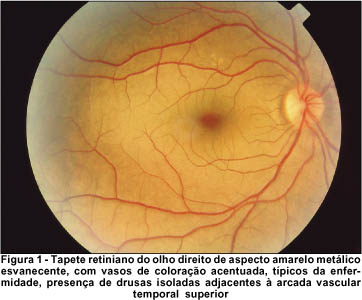
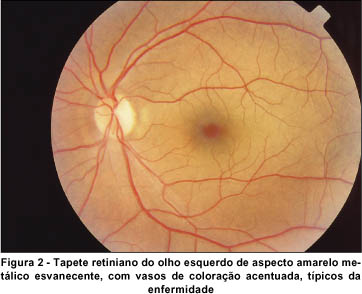
Realizamos o teste para o fenômeno de Mizuo-Nakamura, com oclusão total em AO, em sala escura. Durante 3 horas de adaptação ao escuro, a retina foi re-examinada em intervalos de 1 hora, mas somente após 4 horas não mais apresentava a descoloração peculiar da fundoscopia inicial. O tapete retiniano assumiu coloração e aparência normais, inclusive os vasos sangüíneos perderam sua tonalidade escurecida, quando então repetimos o registro retinográfico (Figuras 3 e 4). Sob exposição à luz do dia, a descoloração peculiar da retina não voltou imediatamente, entretanto, quando checada em intervalos de 2 minutos, foi primeiramente notada após 12 minutos. O fenômeno inteiro foi demonstrado com resultados idênticos noutra ocasião.
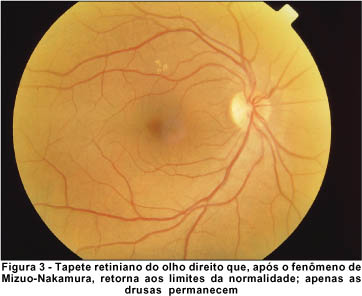
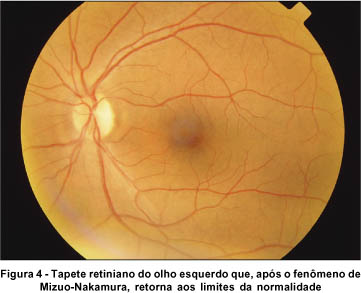
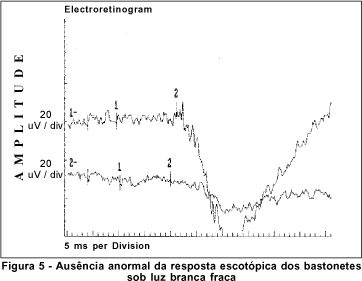
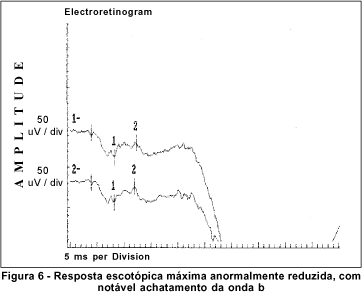
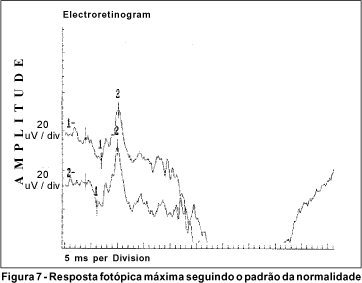
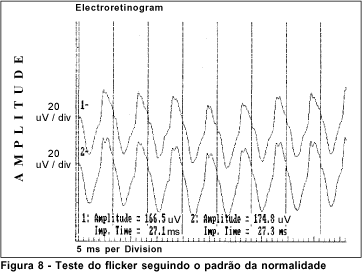
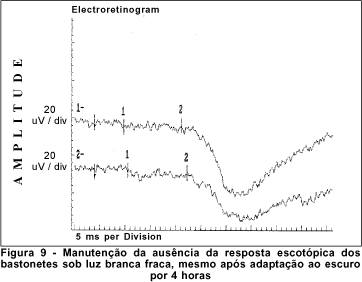
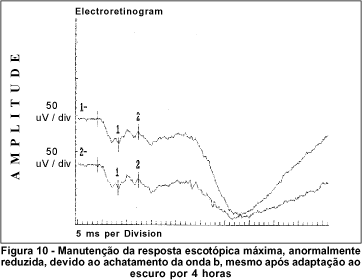
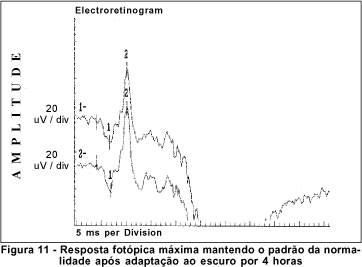
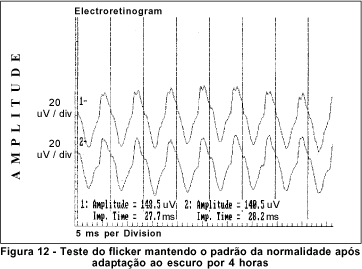
O exame de eletrorretinografia realizado demonstrou ausência de respostas por bastonetes sob luz branca fraca e resposta escotópica máxima sob luz branca forte extremamente reduzida, principalmente no que concerne a onda b. As respostas fotópicas máxima e frente ao flicker foram normais (Figuras 5, 6, 7 e 8). Após adaptação ao escuro por 4 horas, os resultados foram idênticos (Figuras 9, 10, 11 e 12).
DISCUSSÃO
A doença de Oguchi é uma forma incomum de CNCE descrita inicialmente por Chuta Oguchi em 1907(1). Quase que exclusividade do Japão e da Ilha Formosa, onde já foram relatados aproximadamente 140 casos, praticamente não é encontrada no hemisfério ocidental, com algo ao redor de 30 casos relatados(3-4). Como entidade clínica, podemos considerá-la avis rara da patologia oftalmológica(5).
De caráter hereditário autossômico recessivo, a doença aparece acompanhada de história de consangüinidade em aproximadamente 62% dos casos(6). Uma mutação no cromossomo 2q, resultante da deleção da molécula de adenina do nucleotídeo 1147 (1147 delA) no códon 309 do gene responsável pela codificação da proteína arrestina é causa freqüente da doença em pacientes de origem japonesa(7-10). Nos pacientes de ascendência européia, mais recentemente descoberta, a mutação parece decorrer de uma deleção do "exon" 5, no cromossomo 13q, responsável pela codificação da enzima rodopsino-quinase(11-14). Ambas as formas acarretam a codificação de arrestina ou rodopsino-quinase aberrantes que, por suas vezes, falham em desativar a rodopsina para frear a cascata enzimática da foto-transdução, o que foi identificada como causa primária da doença de Oguchi. Por conseguinte, o acúmulo de rodopsina dessensibiliza os bastonetes que só passam a reagir sob limiares maiores. Até que toda a rodopsina seja desativada e "regenerada", num período de 2 a 4 horas, o portador desta doença fica insensível à luz escotópicas, enquanto que no indivíduo normal esta latência é de apenas 10 a 20 minutos(8,14).
Histologicamente, a presença de uma "camada distinta" na retina dos portadores da doença, também presente em pássaros diurnos e certos anfíbios, levou Mann a classificá-la como fenômeno atávico da natureza(15). Investigações posteriores detectaram tal "camada" em autópsias de pacientes não-portadores da doença de Oguchi, o que sugere apenas uma alteração póstuma, bem como também constataram forte correlação anátomo-patológica entre a doença de Oguchi e a retinose pigmentar(16-17).
Com relação ao quadro clínico, raramente existe queixa oftalmológica objetiva, tal como baixa acuidade visual. Entretanto, vale lembrar que a queixa subjetiva de nictalopia pode representar o maior ponto de divergência entre médico e paciente. Há um século, pacientes portadores de cegueira noturna apressavam-se para suas casas logo nos primeiros indícios do pôr-do-sol, a fim de evitar as conseqüências de sua deficiência. Com o advento da iluminação elétrica, veio a possibilidade de ver em cores, usando cones, durante a noite, o que tornou os bastonetes relativamente dispensáveis em várias atividades noturnas. O problema passou a ser despercebido na maior parte das situações, o que faz dos pacientes portadores da doença de Oguchi ou de fundo albipuntacta os mais propensos a ignorar sua condição. Apresentam, portanto, queixa inconsistente e mal-definida(4,8).
Não obstante, algumas características típicas da doença de Oguchi facilitam no diagnóstico diferencial das outras CNCE, sejam fundoscópicas ou eletrofisiológicas. O exame fundoscópico revela alterações bem peculiares que vão de um tapete retiniano de coloração amarelo-metálica a prateada, com um dourado mosqueado, de brilho esvanecente, que se estende das arcadas temporais a médio-periferia da retina, poupando parcialmente o pólo posterior. Os vasos retinianos apresentam-se bastante evidenciados ao contrastarem-se com a coloração retiniana(1,7).
Após aproximadamente 4 horas de adaptação no escuro, a retina lentamente tem todas estas peculiaridades desaparecidas, retornando à absoluta normalidade. A este fenômeno foi dado o nome de Mizuo-Nakamura. Quando da nova exposição à luz, a retina retoma, em prazo de 10 a 20 minutos, todos os comemorativos típicos da doença(2,4,7,18). O fenômeno independe do tipo de iluminação que incide na retina e está relacionado ao acúmulo de potássio extracelular que não pôde ser catabolizado pelas células de Müller que, em última análise, especula-se gerar o reflexo metálico da retina, usualmente observado nesta doença(19-20).
Inicialmente visto como patognomônico da doença, foi isoladamente relatado nas doenças de Stargardt, distrofia de cones recessiva ligada ao X, e retinosquise congênita ligada ao X, embora não tenha sido descrito de maneira tão evidente e duradoura como ocorre na doença de Oguchi onde, ao reverter à condição anormal, mostra latência de 20 a 40 minutos. Nesses relatos isolados, mostrou duração efêmera de apenas alguns segundos(20-21).
Dentre os testes eletrofisiológicos, o eletrorretinograma (ERG) interessa na doença de Oguchi por apresentar, como noutras CNCE, curva anormal de adaptação ao escuro, o que torna o exame crucial no diagnóstico destas doenças. Frente ao teste fotópico máximo e de luz branca a 30 Hz (flicker), tanto o indivíduo normal quanto o portador da doença de Oguchi respondem com traçados normais. Entretanto, frente ao teste escotópico, o portador desta doença responde com traçados anormais, tanto se exposto à luz branca fraca, desencadeadora de resposta escotópica de bastonetes, quanto se exposto à luz branca forte, desencadeadora de resposta escotópica máxima, quando cones e bastonetes são estimulados conjuntamente. Dessa maneira, frente à luz branca fraca, não há qualquer onda (traçado reto) e, frente à luz branca forte, não há resposta de onda b ou esta se apresenta bastante reduzida em amplitude(8).
O eletrorretinograma tem, juntamente com o fenômeno de Mizuo-Nakamura, importância na subclassificação da doença de Oguchi, pois também existem variações dentro do grupo de indivíduos afetados. Três fenótipos são sugeridos: tipo I (típico) - descoloração do fundo, fenômeno de Mizuo-Nakamura com recuperação tardia da adaptação ao escuro; tipo IIA ¾ descoloração do fundo, fenômeno de Mizuo-Nakamura sem recuperação da adaptação ao escuro; tipo IIB ¾ leve descoloração do fundo, ausência ou parcialidade do fenômeno de Mizuo-Nakamura sem recuperação da adaptação ao escuro(22).
CONCLUSÃO
Nosso caso exibiu a descoloração marcante e peculiar da retina e o fenômeno de Mizuo-Nakamura, típicos da doença de Oguchi, porém nenhuma adaptação ao escuro, mesmo após sob prolongada ausência de luz. O reflexo dourado esvanecente da retina, tal qual notado neste paciente, também é menos freqüente do que o cinza-metálico já observado por outros autores(7). Assim, preferimos classificá-lo como portadora da doença de Oguchi tipo II-A. Noutra ocasião seria interessante investigar clinicamente os irmãos desta paciente, possivelmente afetados pela mesma doença.
REFERÊNCIAS
1. Oguchi C. Upon a new night blindness. Acta Ophth Jap 1907;11:123-34.
2. Mizuo G. A new discovery in dark adaptation in Oguchi's disease. Acta Ophth Jap 1913;1148-50.
3. Winn S, Tasman W, Spaeth G, McDonald PR, Justice J. Oguchi's disease in Negroes. Arch Ophthalmol 1969;81:501-7.
4. Sato T, Baba K. Appearance and disappearance of the fundus disturbanc in Oguchi's disease. Am J Ophthalmol 1961;51:243-8.
5. Carr RE, Ripps H. Rhodopsin kinetics and rod adaptation in Oguchi's disease. Invest Ophthalmology 1967;4:426-36.
6. Takagi R, Kawakami R. Uber da Wesen der Oguchischer Krankheit. Klin Monatsbl Augenheilkd 1924;72:349-71.
7. Nakazawa M, Wada Y, Fuchs S, Gal A, Tamai M. Oguchi disease: phenotypic characteristics of patients with the frequent 1147delA mutation in the arrestin gene. Retina 1997;17:17-22.
8. Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol 2000;130:547-63.
9. Maw MA, John S, Jablonka S, Muller B, Kumaramanickavel G, Oehlmann R, et al. Oguchi disease: suggestion of linkage to markers on chromosome 2q. J Med Genet 1995;32:396-8.
10. Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disase in Japanese. Nat Genet 1995;10:360-2.
11. Yamamoto S, Sippel KC, Berson EL, Drayja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997; 15:175-8.
12. Khani SC, Abitbol M, Yamamoto S, Maravic-Magovcevic I, Dryja TP. Characterization and chromosomal localization of the gene for human rhodopsin kinase. Genomics 1996;35:571-6.
13. Khani SC, Nielsen L, Vogt TM. Biochemical evidence for pathogenicity of rhodopsin kinase mutations correlated with the Oguchi form of congenital stationary night blindness. Proc Natl Acad Sci U S A 1998;95:2824-7.
14. Cideciyan AV, Zhao X, Nielsen L, Khani SC, Jacobson SG, Palczewski K. Null mutation in the rhodopsin kinase gene slows recovery kinetics of rod and cone phototransduction in man. Proc Natl Acad Sci U S A 1998;95:328-33.
15. Mann I. Developmental abnormalities of eye. Philadelphia: J B Lippincott; 1957. p.147-9.
16. Kubawara Y, Ishihara K, Akiya S. Histologic and electron microscopic studies of the retina in Oguchi's disease. Jap Rev Clin Ophth 1963;67:1323.
17. Yamanaka M. Histologic study of Oguchi's disease. Its relationship to pigmentary degeneration of the retina. Am J Ophthalmol 1969;68:19-26.
18. Noble KG, Margolis S, Carr RE. The golden tapetal sheen reflex in retinal disease. Am J Ophthalmol 1989;107:211-7.
19. Usui T, Ichibe M, Ueki S, Takagi M, Hasegawa S, Abe H, Sekiya K, Nakazawa M. Mizuo phenomenon observed by scanning laser ophthalmoscopy in a patient with Oguchi disease. Am J Ophthalmol 2000;130:359-61.
20. De Jong PT, Zrenner E, van Meel GJ, Keunen JE, van Norren D. Mizuo phenomenon in X-linked retinoschisis. Pathogenesis of the Mizuo phenomenon. Arch Ophthalmol 1991;109:1104-8.
21. Heckenlively JR, Weleber RG. X-linked recessive cone dystrophy with tapetal-like sheen. A newly recognized entity with Mizuo-Nakamura phenomenon. Arch Ophthalmol 1986;104:1322-8.
22. Carr RE, Gouras P. Oguchi's disease. Arch Ophthalmol 1965;73:646-56.
1 Médico Assistente do Setor de Retina e Vítreo do Departamento de Oftalmologia da Santa Casa de São Paulo. Pós-graduando, nível Doutorado, do Setor de Retina e Vítreo da Clínica Oftalmológica do Hospital das Clínicas da Universidade de São Paulo - USP.
2 Médica Assistente do Setor de Retina e Vítreo do Departamento de Oftalmologia da Santa Casa de São Paulo. Pós-graduanda, nível Mestrado, pela Universidade Federal de São Paulo - UNIFESP.
3 Médica Assistente do Setor de Córnea e Moléstias Externas do Departamento de Oftalmologia da Santa Casa de São Paulo. Pós-graduanda, nível Doutorado, do Setor de Doenças Externas Oculares da Universidade Federal de São Paulo - UNIFESP.
4 Médico Doutor em Medicina, Chefe do Setor de Retina e Vítreo da Clínica Oftalmológica do Hospital das Clínicas da Universidade de São Paulo - USP. Chefe do Setor de Retina e Vítreo do Serviço de Oftalmologia do Hospital Israelita Albert Einstein de São Paulo.
5 Médico Doutor em Medicina pela Universidade de São Paulo - USP de Ribeirão Preto. Chefe do Setor de Retina e Vítreo do Departamento de Oftalmologia da Santa Casa de São Paulo.
Endereço para correspondência: R. Capitão Joaquim da Silva Rocha, 65 ¾ Jundiaí (SP) CEP 13208-750.
E-mail: [email protected]
Recebido para publicação em 18.02.2002
Aceito para publicação em 18.03.2002

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.