Arq. Bras. Oftalmol. 2002; 65 (5): 10.1590/S0004-27492002000500012
Total: 1801
Valdir Balarin Silva1; Paulo Henrique Spirandeli2; Jaqueline Mendonça Lopes de Faria2
DOI: 10.1590/S0004-27492002000500012
RESUMO
Os autores apresentam dois casos de vasculopatia retiniana tipo Coats associada a retinose pigmentar. Estes são os primeiros casos entre irmãos descritos no Brasil e nosso objetivo é anexar mais 2 casos, aos 47 descritos na literatura mundial.
Descritores: Doenças retinianas; Retinite pigmentosa; Consangüinidade; Vasos retinianos; Fotocoagulação; Relato de caso
ABSTRACT
The authors presents two cases of Coats' type exsudative vasculopathy associated with retinitis pigmentosa. These are the first cases in siblings described in Brazil and our purpose is to add two more cases, to the 47 described in the world literature.
Keywords: Retinal diseases; Retinitis pigmentosa; Consanguinity; Retinal vessels; Light coagulation; Case report
INTRODUÇÃO
O termo vasculopatia retiniana exsudativa tipo Coats associada a retinose pigmentar foi recentemente adotado, substituindo as prévias denominações: Doença de Coats, Síndrome de Coats, Descolamento retiniano tipo Coats associado a retinose pigmentar(1-2).
A vasculopatia exsudativa é uma rara ocorrência, tendo sido relatada em 1,2 a 3,6% dos casos de retinose pigmentar avançada(3-5). É usualmente bilateral e não demonstra predileção por sexo. Os achados fundoscópicos são anomalias vasculares telangectásicas na área retiniana inferior afetada, dilatação dos grandes vasos retinianos inferiores já a partir do disco óptico, descolamento retiniano seroso e típicos depósitos de lipídios extra vasculares subretinianos e intraretinianos, multifocais e periféricos; além dos achados típicos relacionados a retinose pigmentar. A retinoangiografia fluoresceínica mostra na periferia retiniana afetada, vasos dilatados, tortuosos e telangectásicos, que extravasam corante. O campo visual é geralmente igual ou menor que 10 graus.
Foi relatada consangüinidade dos pais em quatro casos(6) de associação vasculopatia exsudativa tipo Coats e retinose pigmentar e pouco se sabe sobre uma possível herança genética.
Apenas quarenta e sete casos dessa vasculopatia retiniana específica estão relatados na literatura mundial(7-8). Nosso propósito é anexar mais dois casos, sendo este o primeiro caso entre irmãos relatado no Brasil, até onde temos conhecimento. Anteriormente, apenas sete casos entre irmãos foram relatados na literatura mundial(6,9-10). Discutimos o resultado da fotocoagulação com laser de argônio e a história natural da doença.
RELATO DE CASOS
Caso 1
Paciente de 27 anos, examinada em novembro 1982, sexo feminino, telefonista, pais não consangüíneos, assintomáticos do ponto de vista ocular. Apresentava retinose pigmentar bilateral diagnosticada desde os seis anos de idade. Havia história familiar de hemeralopia em três de sete irmãos, sendo que um desses irmãos não chegou a ser examinado. O exame ocular inicial demonstrou acuidade visual com a melhor correção: 20/100, e menor que J7 em ambos os olhos. A biomicroscopia revelou a presença de opacidade subcapsular posterior do cristalino em ambos os olhos. A fundoscopia confirmou a presença de alterações pigmentares típicas de retinose pigmentar distribuídas por toda a retina até a área peripapilar; disco óptico apresentando elevações arredondadas e isoladas que tornavam suas bordas ligeiramente indistintas, dando aspecto compatível com o diagnóstico clínico de drusas de papila em ambos os olhos, o que é achado relativamente comum na retinose pigmentar. O olho direito apresentava mácula com pigmentação alterada, irregularidade de artérias e veias retinianas no setor inferior, apresentando-se mais dilatadas que as do setor superior, além de tortuosidade acentuada e comunicações artério-venosos (Figura 1). Na região equatorial observava-se descolamento da retina com exsudatos lipídicos no espaço subretiniano. O descolamento retiniano era pouco elevado; estendia-se entre quatro e oito horas. O olho esquerdo demonstrava artérias e veias do setor inferior também dilatadas e tortuosas já a partir do disco óptico, embora em menor grau (Figura 2). Na região equatorial ocorria a presença de alguns exsudatos lipídicos profundos intraretinianos, porém não se observava descolamento de retina. O teste pseudoisocromático HRR, em ambos os olhos, demonstrou ausência total de percepção de cores bilateralmente. O campo visual, pesquisado com mira V-4 no olho direito foi menor que 10º e no olho esquerdo igual a 10º. A ecografia do olho direito confirma o descolamento de retina no setor inferior, sendo que no espaço subretiniano ocorrem ecos espiculares devidos aos lipídeos subretinianos. O exame do disco óptico, em ambos os olhos, com atenuação máxima, demonstrou a persistência de ecos intrapapilares, confirmando o diagnóstico de drusas intrapapilares.
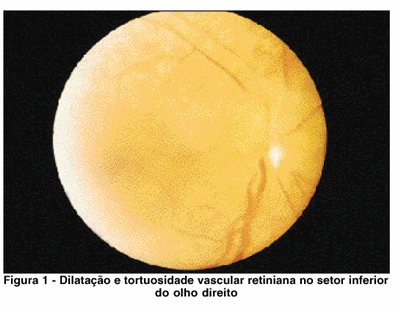
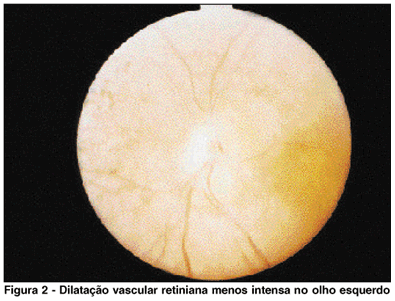
Após tratamento das anomalias vasculares periféricas com laser de argônio em ambos os olhos, a paciente retornou somente após três anos quando se verificou a persistência do descolamento de retina do olho direito, assim como das anomalias vasculares, e notou-se ainda, neovasos epirretinianos na região periférica da retina descolada, com pequena hemorragia vítrea localizada nessa região. O olho esquerdo apresentava aspecto inalterado em relação ao exame anterior. A acuidade visual havia diminuído para 20/200 em virtude de progressão das opacidades cristalinianas em ambos os olhos. Após esta data a paciente não mais retornou.
Caso 2
Paciente de 19 anos, examinada em março de 1983, sexo feminino, auxiliar de escritório, irmã da paciente do caso 1. História de dificuldade visual para longe desde os 10 anos de idade, quando foi prescrita correção óptica. Aos 15 anos, começou a apresentar hemeralopia, entretanto o exame oftalmológico, na ocasião, não detectou alterações retinianas (sic). O exame inicial mostrou acuidade visual com a melhor correção 20/60 e J2 no olho direito e 20/80 e J6 no olho esquerdo. A biomicroscopia não apresentava alterações. A fundoscopia apresentava alterações difusas do epitélio pigmentar com rarefação generalizada e concentrações focais irregulares de pigmento, distribuídos por toda a retina até a região peripapilar, em ambos os olhos. O olho direito apresentava artérias e veias retinianas da região inferior discretamente mais dilatadas que as da região superior; mácula oftalmoscopicamente normal. Na retina periférica inferior, observam-se tortuosidades vasculares. Não foram encontrados no exame comunicações artério-venosos, exsudatos subretinianos ou descolamento de retina. O olho esquerdo mostrava artérias e veias retinianas da região inferior mais dilatadas e tortuosas que no setor superior; disco óptico com bordos ligeiramente indistintos, porém com nítida lobulação, compatível com diagnóstico clínico de drusas de papila (Figura 3); mácula pouco alterada quanto à pigmentação e reflexo foveal. Na região inferior a nível do equador, observavam-se tortuosidades vasculares, comunicações artério-venosos, além de angiomas e retina descolada pouco elevada entre 4 e 8 horas. Observavam-se, no espaço subretiniano, discretos depósitos lipídicos. O teste pseudoisocromático American Optical HRR era alterado em ambos os olhos. O campo visual, pesquisado com mira V-4, foi de 15º em ambos os olhos. Retinoangiografia fluoresceínica, foi realizada e mostrou auto fluorescência típica de drusas intrapapilares, dilatação de vasos inferiores já a partir do disco óptico, presença de grande extravazamento de corante na região dos angiomas retinianos e anomalias vasculares, inferiormente. Nas fases tardias, notou-se ausência de edema cistóide de mácula e presença de tingimento de tecido intrapapilar, confirmando o diagnóstico de drusas.
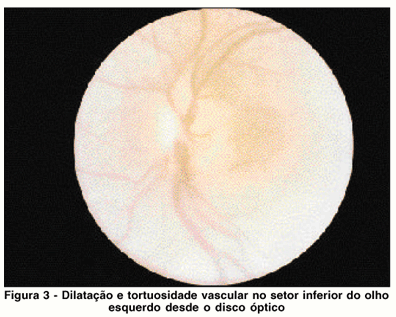
Após o tratamento com laser de argônio das anomalias vasculares pela técnica de disparos seqüenciais, obteve-se ligeira diminuição do calibre desses vasos, entretanto, o descolamento de retina continuou a aumentar, sendo que, quando examinada pela última vez, sete meses após a primeira consulta, apresentava acuidade visual de conta dedos a três metros no olho esquerdo em virtude do descolamento da retina haver atingido a mácula e olho direito mantinha a acuidade visual inicial. Após esta data, a paciente não mais retornou.
DISCUSSÃO
A apresentação desta descrição de casos foi motivada pela pouca freqüência com que tem sido relatada a agora chamada "Vasculopatia retiniana exsudativa tipo Coats associada a retinose pigmentar". Ainda mais raro tem sido o relato da patologia em irmãos, sendo este o oitavo relato na literatura mundial(6,9-10). Retinose pigmentar é sabido ter muitos modos de herança. Na linhagem da família relatada, oito irmãos (quatro homens e quatro mulheres) sendo duas mulheres afetadas, pode ser consistente com retinose pigmentar autossômica recessiva. Small(11) sugeriu que Doença de Coats também pode ser uma anormalidade genética. Nossos achados, de outros autores(6,10) e os de Lanier(9), sugerem a possibilidade da vasculopatia retiniana exsudativa tipo Coats associada a retinose pigmentar ser uma anormalidade com predisposição genética, baseado no fato da ocorrência ser uma patologia rara e estar ocorrendo entre irmãos(10).
Pesquisa recente(12) mostrou que mutações no gene homólogas crumbs (CRB1) são associadas à presença de vasculopatia retiniana exsudativa tipo Coats associada a retinose pigmentar, confirmando assim a natureza genética da associação de patologias.
Diferente da Doença de Coats, que apresenta maior ocorrência em homens no final da primeira década, na forma de uma patologia unilateral, a vasculopatia, associada a retinose pigmentar, apresenta história natural pouco conhecida, não havendo desta forma predileção por sexo ou mesmo idade para o seu início, além de ser binocular.
De uma fase inicial pré-exsudativa, pode evoluir para neovascularização coroideana e retiniana com descolamento crônico da retina, rubeosis iridis com glaucoma neovascular, além de catarata. Esta evolução, nos casos em questão e na literatura mundial, ocorreu em tempo diferente para cada olho no mesmo paciente e pouco dependente ao tratamento realizado.
Pelo fato de não conhecermos exatamente a patogênese desta doença vascular retiniana e o universo a ser analisado ser pequeno, vários métodos de tratamento já foram utilizados e descritos na literatura: fotocoagulação com xenônio(3), laser de argônio, crioterapia(3),introflexão escleral(3). A análise da casuística relatada por outros autores nos leva à conclusão de que os casos avançados são refratários a todas as modalidades terapêuticas.
REFERÊNCIAS
1. Ayesh I, Sanders MD, Friedmann AI. Retinitis pigmentosa and Coats' disease. Br J Ophthalmol 1976;60:775-7.
2. Fogle JÁ, Welch RB, Green WR. Retinitis pigmentosa and exudative vasculopathy. Arch Ophthalmol 1978;96:696-702.
3. Kajiwara Y. Ocular complications of retinitis pigmentosa. Association with Coats' disease. Jpn J Clin Ophthalmol 1980;134:947-955.
4. Khan JA, Ide CH, Strickland MP. Coats' type retinitis pigmentosa. Surv Ophthalmol 1988;32:317-32.
5. Pruett RC. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc 1983;81:693-735.
6. Harada T, Yasuma M, Ichikawa H. Coats' syndrome hemorrhages. Rinsho Ganka 1978;32:1629-36.
7. Fortes FJB, Nehemy MB. Retinose pigmentar associada com vasculopatia exsudativa periférica retiniana. Rev Bras Oftalmol 1996;55:39-46.
8. Abu El-Aspar AM, Kahtani ES, Tabbara KF. Retinal arteriovenous communication in retinitis pigmentosa with Refsum's disease-like findings. Doc Ophthalmol 1995;89:313-20.
9. Lanier JD, McCrary JA, Justice J. Autossomal recessive retinitis pigmentosa and Coats' disease: a presumed familial incidence. Arch Ophthalmol 1976; 94:1736-42.
10. Kollarits RC, Mehelas TJ, Shealy TR, Zahn JR. Von Hippel tumors in siblings with retinitis pigmentosa. Ann Ophthalmol 1982;14:256-9.
11. Small RG. Coats' disease and muscular dystrophy. Trans Am Acad Ophthalmol Otolaryngol 1968;72:225-31.
12. Den Hollander AI, Heckenlively JR, Van den Born LI, Kok YJM, Van der Velde-Visser SD, Kellner U, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) Gene . Am J Hum Genet 2001;69:198-203.
1 Professor Assistente Doutor de Oftalmologia do Departamento de Oftalmologia e Otorrinolaringologia da Universidade Estadual de Campinas - UNICAMP.
2 Pós-Graduando nível Mestrado em Oftalmologia pela Faculdade de Ciências Médicas da Universidade Estadual de Campinas - UNICAMP.
3 Professora participante da Pós-Graduação da Faculdade de Ciências Médicas da Universidade Estadual de Campinas - UNICAMP.
Endereço para correspondência: Rua Seis, 1353 - Rio Claro (SP) CEP 13500-190.
E-mail: [email protected]
Recebido para publicação em 20.04.2001
Aceito para publicação em 30.01.2002
Nota editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos à Dra. Luciana Peixoto Finamor Santos.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.