Arq. Bras. Oftalmol. 2002; 65 (2): 10.1590/S0004-27492002000200004
Total: 1449
Rosane da Cruz Ferreira1; Faye Oelrich2; Bronwyn Bateman3
DOI: 10.1590/S0004-27492002000200004
ABSTRACT
Purpose: To evaluate the genetic aspects of strabismus. Methods: Ophthalmic and orthoptic evaluations were performed prospectively on 110 strabismic probands and 478 relatives. We used 3 different criteria in the diagnosis of strabismus: primary diagnosis (dx1) defined as any manifest horizontal or vertical deviation, a secondary diagnosis (dx2) including esophoria (>7 prism diopters) or exophoria (>9 prism diopters), and a tertiary diagnosis (dx3) including abnormal fusional amplitudes, accommodative convergence/accommodation (AC/A) ratio, and/or stereopsis; monofixation syndrome; 4 prism diopters base out; and/or abnormal Maddox test responses. Analyses were carried out within mating types. Results: Hypotheses of autosomal dominant or recessive inheritance with no sporadics were rejected. Based on the dx1, 25% of the families had more than one individual affected and there was vertical transmission in 13%; adding dx2 there were 36% of the families with more than one affected and 21% had vertical transmission; and adding dx3, there were 73% with more than one affected and 51% with vertical transmission. Conclusions: There is evidence for a pattern consistent with an autosomal dominant form of strabismus in most families.
Keywords: Strabismus; Esotropia; Exotropia; Binocular vision; Orthoptics
RESUMO
Objetivo: Avaliar aspectos genéticos em estrabismo. Métodos: Foram realizados exames oftalmológicos e ortópticos prospectivamente em 110 propósitos estrábicos e 478 familiares. Foram usados 3 diferentes critérios no diagnóstico de estrabismo: diagnóstico primário (dx1) foi definido como qualquer desvio horizontal ou vertical manifesto, diagnóstico secundário (dx2) incluiu esoforias (> 7 dioptrias prismáticas) ou exoforia (< 9 dioptrias prismáticas) e diagnóstico terciário (dx3), que incluiu amplitudes de fusão anormal, relação convergência acomodativa/acomodação (CA/A) elevada, estereopsia anormal, síndrome de monofixação, e teste das 4 dioptrias prismáticas e teste de Maddox anormais. A análise dos resultados foi por meio da avaliação dos pedigrees segundo o tipo de união. Resultados: As hipóteses de herança autossômicas dominantes ou recessivas sem casos esporádicos foram rejeitadas. Baseado no dx1, 25% das famílias tiveram mais de um indivíduo afetado e houve transmissão vertical em 13%; adicionando o dx2, foi encontrado que 36% das famílias tinham mais de um membro afetado e houve transmissão vertical em 21%; e adicionando dx3, 73% das famílias tinham mais de um membro afetado e houve transmissão vertical em 51% delas. Conclusão: Existem evidências que apontam para um padrão compatível com herança autossômica dominante na maioria das famílias.
Descritores: Estrabismo; Esotropia; Exotropia; Visão binocular; Ortóptica
INTRODUCTION
Strabismus occurs in 1.3 to 5.7% of all children (1-3). The pathogeneses of strabismus are unknown and the precise mode(s) of inheritance is still obscure (4-9). Frequently, the criteria for assignment of affected status have been poorly defined and/or the historical data with inherent inaccuracies. In addition, conclusions have been drawn from individual pedigrees (4-5).
There is no statistical difference in the prevalence of strabismus be-tween genders (2) and the most common type of manifest comitant strabismus is esotropia, occurring approximately 3 times more often than exotropia (6).
The coexistence of different types of strabismus (exotropia and esotropia) within the same family has been well documented (4,7,9-12). Monozygotic twins may have different forms of strabismus (12) and families with both exotropia and esotropia have been reported (4,9,11). Some investigators have postulated that there may be a common pathogenic factor that results in a variety of ocular motility disturbances either convergent or divergent (3).
This study was undertaken to prospectively investigate the genetic aspects of strabismus in a reasonable number of families sampled in a systematic way with consistent clinical definitions.
METHODS
Probands (Jules Stein Eye Institute, UCLA, Los Angeles, CA) were consecutively selected if they had any manifest horizontal and/or vertical deviation and no restriction or paralysis of extraocular muscles. Inclusion criteria were normal physical, motor, neurological and mental function; probands with known genetic syndromes were excluded. After approval by the UCLA Human Subject Protection Committee and obtained informed consent, complete ophthalmic and orthoptic evaluations were performed on probands and all available relatives by the same person (FO). Evaluation consisted of visual acuity, measurements of deviations at distance and near with the cover-uncover test, alternate cover test with prisms or Krimsky test, fusional amplitudes, accommodative-convergence/accommodation (AC/A) ratio determination, stereopsis test ( Titmus test - Stereo Optical Co, Inc, Chicago, Il), 4 D prism diopter base out test, Maddox test, and cycloplegic refraction with cyclopentolate. Some tests were not possible in some subjects because of inadequate cooperation.
Tests of fusional amplitudes were made using the Risley (13) rotatory prism and the values considered abnormal are the following: (a) prism convergence break point at 20 feet for male if < 5.1 or >32 D and for female <6.7 or >32; (b) prism divergence break point at 13 inches for male if <12.2 or >29.4 D and for female <12.1 or > 29.3; and (c) prism divergence break point at 20 feet for male <1.1 or > 12.7 and for female <1.5 or >13.9 D (11,14-15). The AC/A ratio was measured with the slope-gradient method using a -1.5 and a -3.00 lens (16-17); an AC/A ratio greater than 5 was considered abnormal for both males and females (18-19). Stereopsis was considered abnormal if less than 100 sec (20) and not included in the analyses in individuals younger than 5 years. The 4 D diopter base out test was done using a single 4 diopter prism in front of each eye separately and considered abnormal if microstrabismus or absence of bifoveal fixation was present (5). Maddox test was abnormal if there was any tortional strabismus (21).
Analyses were performed with progressive groups defining "affected" status. Initially, relatives were considered as "affected" if they had any manifest horizontal and/or vertical deviation (primary diagnosis = dx1). Secondary diagnosis (dx2) included those individuals with dx1 and those with esophoria greater than 7 prism diopters or exophoria greater than 9 (13-14,17-18). Tertiary diagnosis (dx3) additionally included individuals with abnormal fusional amplitudes, AC/A ratio, stereopsis, 4 D diopter base out test and/or Maddox test.
Analyses of the pedigrees were carried out within mating type. The mating type of the proband's parents was NxN if both were unaffected, NxA if one parent was normal, the other affected, and AxA when both were affected. Some individuals had incomplete examinations and affected status could not be assigned. Those individuals were considered as "unknown" (U), thus, originating matings NxU, AxU and UxU. These pedigree analyses based on the prevalence of males and females affected and the existence of male to male transmission allow to exclude or not X-linked inheritance.
Dx2 was used to study the different types of strabismus in members of the same family. Dx3 included other sensory (abnormal stereopsis, absence of bifoveal fixation) and motor (abnormal fusional amplitudes, abnormal Maddox test) abnormalities; subtyping of affected relatives into eso- and exotropias was not possible.
RESULTS
One hundred and ten probands identified 107 families. Three families had 2 probands; 329 relatives had complete examinations and an additional 149 had some missing data ("unknown" status).
There were 63 (56.4%) females and 48 (43.6%) male probands; this does not differ from the standard 1:1 ratio ( X 2 = 2.3; 1 dF). The ages ranged from less than one year to 48 years. Thirty eight (34.6%) of the probands had exotropia, 70 (63.6%) had esotropia, and 2 (1.8%) had vertical deviation only. Among the esotropias, 35 (50%) were infantile esotropia, 26 (37.2%) had a spherical equivalent greater than 1.5 diopters and/or abnormal AC/A ratio and 9 (12.8%) were other types of esotropia.
Consanguinity was not identified in any family.
Among 329 relatives with complete examinations, 28 (8.5%) had horizontal tropia, or vertical tropia or phoria and were considered affected (dx1). Adding the secondary diagnosis (dx2), 15 more individuals were affected - total of 43 people (13%); including dx3, an additional 78 people were included as affected - total of 121 individuals (36.8%).
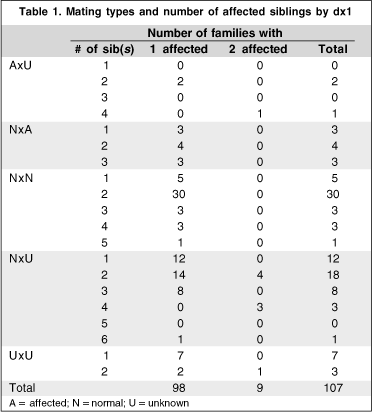
Using dx1, 27 (25.2%) of 107 families had more than one individual affected; 9 (8.4%) of the siblings of the probands were affected ( Table 1); there was vertical transmission in 14 (13%) and male to male transmission occurred in 5 families. Using dx2, 39 (36.4%) of the families had more than one individual affected; 11 (10.3%) of the siblings of the probands were affected ( Table 2); there was vertical transmission in 23 families (21.5%) and male to male transmission occurred in 13. Using dx3, 78 (72.9%) of the families had more than one individual affected; 21 (19.6%) of the siblings of the probands were affected ( Table 3); there was vertical transmission in 55 families (51.4%) and male to male transmission in 21 ( Figure 1). The proportion of family members considered affected rose dramatically across the 3 diagnoses. Using dx1, 28 (8.5%) of 329 relatives were considered affected; adding dx2, 43 (13%) were affected; and adding dx3, 121 (36.8%).



The AC/A ratio was abnormal in 3.5% family members; Titmus test was abnormal in 5.1% relatives; and the 4 D prism diopter base out test was abnormal in 1.7% family members. Fusional amplitudes were measured in 280 relatives and were abnormal in 91 (32.5%). In our study, prism convergence break point at 13 inches was not used because its measurements are quite variable. The ability to convert at near can be learned and improved with training and the use of these values in this research could lead to inaccurate results (15).
Thirty-nine families had more than one individual affected by dx2 and these individuals had either eso- or exotropias, or eso- or exophorias. Of 39 families with more than one member affected, 9 (23.1%) had esotropia in all affected members, 12 (30.8%) had exotropia in all affected and 18 (46.1%) of the families had both types in affected members ( Figure 1).
DISCUSSION
The inconclusive and often conflicting results of previous studies are in part a result of problems inherent in the diagnosis of strabismus (22-24). Genetic analyses require unambiguous classification of family members, as normal or abnormal, and unfortunately, suitable criteria for the diagnosis of strabismus are lacking. Strabismus may be considered as any manifest deviation, esophorias greater than 7 at distance or near, and exophorias greater than 9 prism diopters at distance or at near; we used these criteria (14). The US National Health Survey (2) defined abnormal phorias as "deviations of the binocular fixation point in the horizontal plane at a distance of 5 prism diopters or more of convergence (significant esophoria) and 5 prism diopters or more of divergence (significant exophoria)"; at near, "deviations of 6 prism diopters or more convergence were considered significant esophoria, and 10 prism diopters or more of divergence, significant exophoria".
Since Mendelian laws were established, authors have been trying to establish strabismus inheritance (4,8-9,11,22). Autosomal dominance with reduced penetrance was advocated for primary concomitant esotropia because the strabismus was not observed in each generation (22). In 1923, a recessive mode of inheritance was proposed for esotropia based on the study of 306 families; however, the author failed to find the expected 25 per cent of strabismic offspring (23). In 1954, it was suggested that there is an "irregular" dominance in strabismus which the author attributed to reduced penetrance and variable expressivity. It was postulated that there "might be a dominant chief gene, supported by mainly dominant accessory genes" (4).
There is a continuum of measurable abnormalities within strabismus from heterophoria, through intermittent tropia to manifest strabismus (4,10). Abnormal sensory and motor tests have been found to be statistically more frequent among relatives of a strabismic child than among relatives of normal children (13,15,18-20). Fusional amplitudes of parents of normal, esotropic and exotropic children were studied (16). Parents of the esotropic and exotropic groups were found to have lower values of fusional amplitudes than parents of normal children. In our study almost one third (32.5%) of the relatives of the proband were found to have abnormal fusional amplitudes.
In 1967 the first to attempt was made to determine the inheritance of strabismus based on modern genetic methodology (9). 697 probands with eso- or exotropias and their available relatives were studied and it was found that the number of siblings of the proband with strabismus depended upon whether or not the parents were affected. The author reported that 30 to 50% of the siblings of patients with strabismus exhibited strabismus if one or both parents were affected, and 20 to 30% if both parents were unaffected. There was an increase in approximately 5% in all these figures if heterophoria (for her purposes considered to be a deviation of more than 4 degrees) was included. Her study was similar to ours as she assessed the binocular vision status of all available family members including the apparently normal relatives. She proposed a multifactorial inheritance based on close evaluation of quantitative factors such as heterophoria and deficiencies in binocular vision in relatives of strabismic children.
In 1986, a homogeneous group of infantile (congenital) esotropia patients diagnosed prior to the age 6 months (in the absence of refractive errors of more than +1.50 diopters spherical equivalent) was studied (7). Using segregation analyses, families of probands were studied and compiled a total of 173 pedigrees with 1589 people. The criteria for affected relatives was not specified. The results of this study supported a Mendelian codominant model, however, the estimated transmission probability for this codominant model was substantially different from the Mendelian expectations, suggesting the existence of etiologic heterogeneity among families, which would result from admixture of a major proportion of autosomal recessive cases, some dominant cases, and possibly aggregation of nongenetic cases. Their sample size was larger than ours; however, they did not examined everyone in their sample, and most relatives were considered affected or not based on historical data only. They did not consider possible manifestations of a gene for infantile esotropia such as dissociated vertical deviation or reduced binocularity in the relatives who were presumably normal.
Recently, family history was confirmed as a risk factor for strabismus (25). Family history is significantly more prevalent in intermitent exotropia and accommodative or partially accommodative esotropia.
We studied a reasonable number of families sampled in a systematic way with consistent clinical definitions, to evaluate the genetic aspects of strabismus. We found a high prevalence of different types of strabismus within the same family. Almost half (46%) of the families with more than one individual affected had both eso- and exodeviations in their members. Three diagnostic categories (dx1, dx2 and dx3) were used in this study to identify affected individuals based on abnormal sensory or motor ocular status. By using inclusive criteria for assigning affected status, the prevalence of familial cases and the number of affected members in each family increased from 25.2% to 72.9% and from 8.5% to 36.8% respectively. There could have been some distortion since young people who could not cooperate in some of the tests used in the dx3 were coded as "unknown".
The lack of statistical difference between genders and the presence of male to male transmission excluded X-linked inheritance as a possible inheritance pattern in our population. The high prevalence of vertical transmission (see for example one of our three generation pedigrees, Figure 1) and absence of consanguinity strongly suggest a dominant mode of inheritance, with some sporadic cases. However, for common disorders the distinction between dominant and recessive is difficult to establish because the segregation ratios differ only for some mating types.
The parsimonious explanation of the inheritance of strabismus in our study, using our 3 diagnoses and considering the total group, is a high percentage of sporadic cases and a familial form of strabismus in which the gene and/or genes are inherited as an autosomal variant.
REFERENCES
1. Bateman JB, Isenberg SJ. Genetic aspects of strabismus. In: Principles and Practice of Medical Genetics . 2 nd ed. New York: Churchill Livingstone; 1990. p.723-31.
2. National Center for Health Statistics 1972. Eye Examination Findings Among Children . Washington DC: US Department of Health, Education and Welfare; 1972. p.16-42.
3. Podgor MJ, Remaley NA, Chew E. Associations between siblings for esotropia and exotropia. Arch Ophthalmol 1996;114:739-44.
4. Waardenburg PJ. Squint and heredity. Doc Ophthalmol 1954;7:422-94.
5. Cantolino SJ, von Noorden GK. Heredity in microtropia. Arch Ophthalmol 1969;81:753-7.
6. Mash AJ, Spivey BE. Genetic aspects of strabismus. Doc Ophthalmol 1973;34:285-91.
7. Maumenee IH, Alston A, Mets MB, et al. Inheritance of congenital esotropia. Trans Am Ophthalmol Soc 1986;84:85-93.
8. Paul TO, Hardage LK. The heritability of strabismus. Ophthalmic Genet 1994;15:1-18.
9. Richter S. Zur Heredität des strabismus concomitans. Humangenetik 1967;3:235-43.
10. Grützner IP, Yazawa K, Spivey BE. Heredity and strabismus Surv Ophthalmol 1970;14:441-56.
11. Schlossman A, Priestley EL. Relationships between lateral heterophoria, prism vergence, and the near point of convergence. Am J Ophthalmol 1948;31:427-41.
12. Devries B, Houtman WA. Squint in monozygotic twins. Doc Ophthalmol 1979;46:305-8.
13. Scobee RG, Green EL. Tests for heterophoria. Am J Ophthalmol 1947;30:436-51.
14. Graham PA. Epidemiology of strabismus. Br J Ophthalmol 1974;58:224-31.
15. Scobee RG, Green EL. Relationships between lateral heterophoria, prism vergence, and the near point of convergence. Am J Ophthalmol 1947;31:427-41.
16. Niederecker O, Mash AJ, Spivey BE. Horizontal fusional amplitudes and versions. Comparison in parents of strabismic and nonstrabismic children. Arch Ophthalmol 1972;87:283-5.
17. Laatikainen L, Erkkila H. Refractive errors and other ocular findings in school children. Acta Ophthalmol 1980;58:129-36.
18. Mash AJ, Hegmann JP, Spivey BE. Genetic analysis of cover test measures and AC/A ratio in human populations with varying incidences of strabismus. Brit J Ophthalmol 1975;59:380-4.
19. Franceschetti AT, Burian HM. Gradient accommodative convergence/accommodative ratio in families with and without esotropia. Am J Ophthalmol 1970;70:558-62.
20. Smith D, Grützner P, Colenbrander A, Hegmann JP, Spivey B. Selected ophthalmologic and orthoptic measurements in families. Arch Ophthalmol 1972;87:278-82.
21. Snydacker D. The Maddox rod test: a ten year follow-up. Am J Ophthalmol 1963;55:278-82.
22. François J. Affections of the ocular muscles. In: Heredity in Ophthalmology, CV Mosby St Louis; 1961. p.239-69.
23. Czellitzer A. Wie vererbt sich Schielen. Arch Rassen Gesellsch Biol 1923;14:377-94.
24. Abrahamsson M, Magnusson G, Sjöstrand J. Inheritance of strabismus and the gain of using heredity to determine populations at risk of developing strabismus. Acta Ophthalmol Scand 1999;77:653-7.
25. Matsuo T, Yamane T, Ohtsuki H. Heredty versus abnormalities in pregnancy and delivery as risk factors for different types of comitant strabismus. J Pediatr Ophthalmol Strabismus 2001;38:78-82.
1
Department
of
Ophthalmology,
Federal
University
of
São
Paulo,
Brazil
and
Department
of
Ophthalmology,
Rocky
Mountain
Lions
Eye
Institute,
The
Children's
Hospital,
University
of
Colorado,
Denver,
Colorado.
2
Department
of
Ophthalmology,
Jules
Stein
Eye
Institute,
UCLA,
Los
Angeles,
California.
3
Department
of
Ophthalmology,
Rocky
Mountain
Lions
Eye
Institute,
The
Children's
Hospital,
University
of
Colorado,
Denver,
Colorado.
Corresponding
author:
Rua
Maracá,
70
¾
Porto
Alegre
(RS)
CEP
91900-640.
E-mail:
[email protected]
Nota
Editorial:
Pela
análise
deste
trabalho
e
por
sua
anuência
sobre
a
divulgação
desta
nota,
agradecemos
aos
Drs.
José
Paulo
Cabral
Vasconcellos
e
Henderson
Celestino
de
Almeida.
Recebido
para
publicação
em
01.03.2001
/
Aceito
para
publicação
em
01.10.2001

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.