Arq. Bras. Oftalmol. 2012; 75 (5): 10.1590/S0004-27492012000500012
Total: 2792
Luciano Sólia Nasser1; Lívia Maris Ribeiro Paranaíba1; Ana Cláudia Frota2; Andreia Gomes2; Gisele Versiani3; Hercílio Martelli Júnior1
DOI: 10.1590/S0004-27492012000500012
ABSTRACT
PURPOSE: To describe the clinical and imaginological features of two families with Waardenburg syndrome: type I and II, with emphasis on ophthalmic manifestations, as well as the pattern of genetic inheritance. METHODS: We conducted a clinical study involving two families affected by Waardenburg syndrome, and through the pedigree, determined the present pattern of genetic inheritance. Analyses were performed including the measurement of visual acuity, the presence of dystopia cantorum (telecanthus), evaluation of iris color and retinal mapping, as well as dermatological and otological examinations. RESULTS: The pedigree of the family affected by the Waardenburg syndrome type I showed an autosomal dominant mode of transmission. The syndrome was present at 85.71% of patients. The dystopia cantorum was the most frequent feature, followed by the white streak on the skin of the forehead, hypopigmentation of the iris and retina and deafness. The Waardenburg syndrome family type II had 33.33% of family members affected by the syndrome. No member had dystopia cantorum and hypopigmentation of the iris. Three patients had sensorineural hearing loss (12.5%), associated with white forelock and achromatic spots confluent by the body. CONCLUSION: This study shows the importance of the ophthalmologist in aiding the diagnosis of this rare genetic condition, since it includes ocular disorders such as telecanthus, hypopigmentation of the iris and retina. The cantorum dystopia is the main diagnostic criterion to differentiate type I and II syndrome and should be done by a trained ophthalmologist. The families are in medical monitoring, receiving genetic guidelines and care related to eye protection.
Keywords: Waardenburg's syndrome; Iris diseases; Retinal diseases; Genetic counseling; Deafness; Eyelids; Nose; Case reports
RESUMO
OBJETIVO: Descrever as características clínicas e imaginológicas de duas famílias com a síndrome de Waardenburg, sendo uma do tipo I e outra do tipo II, enfatizando as manifestações oftalmológicas, bem como o padrão de herança genética. MÉTODO: Realizou-se um estudo clínico envolvendo as duas famílias afetadas pela síndrome de Waardenburg, sendo, através dos heredogramas, determinado o padrão de herança genética presente. Também foram realizadas análises oftalmológicas abordando a medida da acuidade visual, a presença de distopia cantorum (telecanto), a avaliação da coloração da íris e o mapeamento de retina, além de exames otológicos e dermatológicos. RESULTADOS: O heredograma da família afetada pela síndrome de Waardenburg tipo I revelou um modo autossômico dominante de transmissão. A condição estava presente em 85,71% dos pacientes. A distopia cantorum foi a alteração mais frequente, seguida pela mecha branca na pele da fronte, hipopigmentação da íris e da retina e surdez neurossensorial. A família com síndrome de Waardenburg tipo II apresentou 33,33% dos familiares com a alteração. Nenhum membro apresentou distopia cantorum e hipopigmentação de íris. Três pacientes apresentaram surdez neurossensorial (12,5%), associada ao topete branco e manchas acrômicas confluentes pelo corpo. CONCLUSÃO: O presente estudo mostra a importância do oftalmologista no auxílio do diagnóstico desta rara condição genética, uma vez que inclui alterações oftalmológicas como telecanto, hipopigmentação da íris e retina. A distopia cantorum é o principal critério diagnóstico para diferenciar o tipo I do II e deve ser feita por oftalmologista treinado. As famílias encontram-se em acompanhamento multiprofissional, tendo recebido orientações genéticas e os cuidados referentes à proteção ocular.
Descritores: Síndrome de Waardenburg; Doenças da íris; Doenças retinianas; Aconselhamento genético; Surdez; Pálpebras; Nariz; Relatos de casos
RELATOS DE CASOS CASE REPORTS
Síndrome de Waardenburg - aspectos oftalmológicos e critérios de diagnóstico: relatos de casos
Waardenburg syndrome - ophthalmic findings and criteria for diagnosis: case reports
Luciano Sólia NasserI; Lívia Maris Ribeiro ParanaíbaI; Ana Cláudia FrotaII; Andreia GomesIII; Gisele VersianiIV; Hercílio Martelli JúniorI
IPrograma de Pós-graduação em Ciências da Saúde, Universidade Estadual de Montes Claros, Unimontes, Montes Claros, Minas Gerais, Brasil
IIEstudante de Medicina, Universidade Estadual de Montes Claros, Unimontes, Montes Claros, Minas Gerais, Brasil
IIIEstudante de Odontologia, Universidade Estadual de Montes Claros, Unimontes, Montes Claros, Minas Gerais, Brasil
IVDepartamento de Fonoaudiologia do Hospital Universitário Clemente de Faria da Universidade Estadual de Montes Claros, Unimontes, Minas Gerais, Brasil
RESUMO
OBJETIVO: Descrever as características clínicas e imaginológicas de duas famílias com a síndrome de Waardenburg, sendo uma do tipo I e outra do tipo II, enfatizando as manifestações oftalmológicas, bem como o padrão de herança genética.
MÉTODO: Realizou-se um estudo clínico envolvendo as duas famílias afetadas pela síndrome de Waardenburg, sendo, através dos heredogramas, determinado o padrão de herança genética presente. Também foram realizadas análises oftalmológicas abordando a medida da acuidade visual, a presença de distopia cantorum (telecanto), a avaliação da coloração da íris e o mapeamento de retina, além de exames otológicos e dermatológicos.
RESULTADOS: O heredograma da família afetada pela síndrome de Waardenburg tipo I revelou um modo autossômico dominante de transmissão. A condição estava presente em 85,71% dos pacientes. A distopia cantorum foi a alteração mais frequente, seguida pela mecha branca na pele da fronte, hipopigmentação da íris e da retina e surdez neurossensorial. A família com síndrome de Waardenburg tipo II apresentou 33,33% dos familiares com a alteração. Nenhum membro apresentou distopia cantorum e hipopigmentação de íris. Três pacientes apresentaram surdez neurossensorial (12,5%), associada ao topete branco e manchas acrômicas confluentes pelo corpo.
CONCLUSÃO: O presente estudo mostra a importância do oftalmologista no auxílio do diagnóstico desta rara condição genética, uma vez que inclui alterações oftalmológicas como telecanto, hipopigmentação da íris e retina. A distopia cantorum é o principal critério diagnóstico para diferenciar o tipo I do II e deve ser feita por oftalmologista treinado. As famílias encontram-se em acompanhamento multiprofissional, tendo recebido orientações genéticas e os cuidados referentes à proteção ocular.
Descritores: Síndrome de Waardenburg; Doenças da íris; Doenças retinianas; Aconselhamento genético; Surdez; Pálpebras/anormalidades; Nariz/anormalidades; Relatos de casos
ABSTRACT
PURPOSE: To describe the clinical and imaginological features of two families with Waardenburg syndrome: type I and II, with emphasis on ophthalmic manifestations, as well as the pattern of genetic inheritance.
METHODS: We conducted a clinical study involving two families affected by Waardenburg syndrome, and through the pedigree, determined the present pattern of genetic inheritance. Analyses were performed including the measurement of visual acuity, the presence of dystopia cantorum (telecanthus), evaluation of iris color and retinal mapping, as well as dermatological and otological examinations.
RESULTS: The pedigree of the family affected by the Waardenburg syndrome type I showed an autosomal dominant mode of transmission. The syndrome was present at 85.71% of patients. The dystopia cantorum was the most frequent feature, followed by the white streak on the skin of the forehead, hypopigmentation of the iris and retina and deafness. The Waardenburg syndrome family type II had 33.33% of family members affected by the syndrome. No member had dystopia cantorum and hypopigmentation of the iris. Three patients had sensorineural hearing loss (12.5%), associated with white forelock and achromatic spots confluent by the body.
CONCLUSION: This study shows the importance of the ophthalmologist in aiding the diagnosis of this rare genetic condition, since it includes ocular disorders such as telecanthus, hypopigmentation of the iris and retina. The cantorum dystopia is the main diagnostic criterion to differentiate type I and II syndrome and should be done by a trained ophthalmologist. The families are in medical monitoring, receiving genetic guidelines and care related to eye protection.
Keywords: Waardenburg's syndrome; Iris diseases; Retinal diseases; Genetic counseling; Deafness; Eyelids/abnormalities; Nose/abnormalities; Case reports
INTRODUÇÃO
Síndrome de Waardenburg (SW; MIM #193500) é uma alteração do sistema melanocítico-pigmetário resultante de uma desordem na diferenciação, sobrevivência e migração dos melanócitos derivados da crista cristal no período embriogênico(1). A SW apresenta distribuição universal, sem predileção por raça e gênero e sua prevalência estimada é de 1:42.000 nativivos na população geral(2).
Análise histológica das estruturas afetadas na SW mostra ausência de melanócitos teciduais como na região frontal do cabelo (topete branco), pele, íris e estria vascular da cóclea (surdez neurosensorial)(3). Além destas alterações de pigmentação, pode ocorrer a distopia cantorum ou telecanto, um deslocamento lateral dos cantos internos dos olhos(2). Características menos frequentes associadas são, a presença de manchas acrômicas da pele, a confluência dos supercílios (sinófris) e uma base larga do nariz(4). Diante desta diversidade clínica, foram propostos os critérios diagnósticos maiores para a SW: perda auditiva neurossensorial, alterações na pigmentação da íris, hipopigmentação do cabelo (topete branco ou cabelos grisalhos surgidos antes dos 30 anos de idade) e distopia cantorum, além de critérios menores: sinófris, hipopigmentação da pele e uma base larga do nariz(4-8). Para ter a confirmação da SW o paciente deve apresentar dois critérios maiores e um menor(4).
De acordo com a combinação dessas alterações clínicas, a SW é classificada em 4 tipos, SW1, SW2, SW3 e SW4. O tipo I (SW1; MIM #193500) é a forma clássica com distopia cantorum, o tipo II (SW2; MIM #193510) caracterizado pela presença de tecidos com distúrbios de pigmentação e surdez, porém, sem a presença de distopia cantorum, o tipo III (síndrome de Klein-Waardenburg; MIM #148820) é similar ao tipo I com anormalidades musculoesqueléticas adicionais; enquanto o tipo IV (síndrome de Shah-Waardenburg ou doença de Waardenburg-Hirschsprung, MIM #277580) é caracterizada pela presença de megacolon congênito(4). A tabela 1 mostra os critérios de diagnóstico para a SW.

O objetivo do presente estudo foi descrever as características clínicas e imaginológicas em 2 famílias brasileiras com a SW (tipo I e tipo II), enfatizando as manifestações oftalmológicas.
MÉTODOS
Família 1
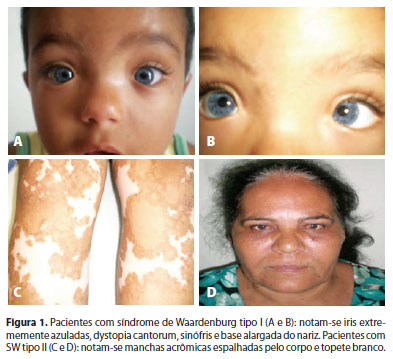
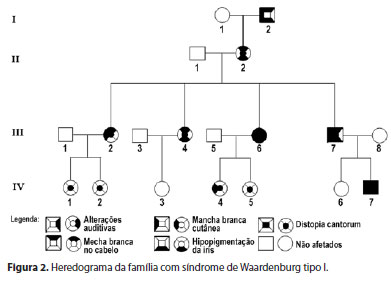
Paciente de 2 anos, masculino, procedente de Montes Claros, Minas Gerais, acompanhado de seus pais, apresentava surdez total bilateral desde o nascimento. Foi avaliado no departamento de oftalmologia da Universidade Estadual de Montes Claros, Unimontes, Minas Gerais, Brasil, por apresentar queixa principal de fotofobia e os olhos extremamente claros. Ao exame clínico, observou-se uma mecha branca na região cutânea da fronte, uma mancha hipocrômica no braço direito, íris extremamente azuis e telecanto (Figura 1). Diante da clínica observada, o diagnóstico presuntivo foi de SW. Iniciou-se um estudo envolvendo toda a família do paciente, composta por 15 membros nas últimas quatro gerações, para determinar o padrão de herança e a expressividade da doença (Figura 2).
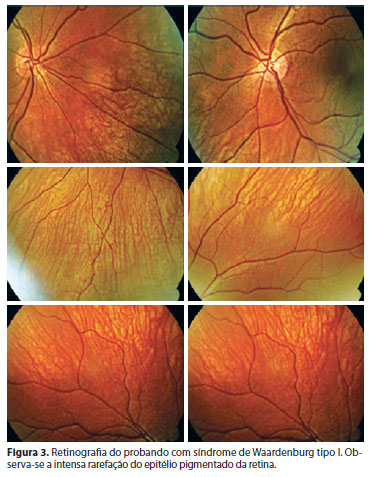
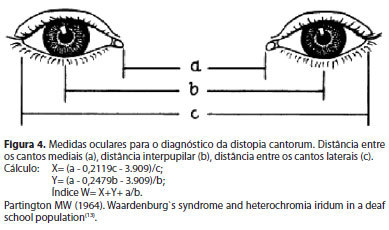
Os pacientes afetados tinham idade entre 2 e 55 anos (média de 18,27 anos). A avaliação clínica incluiu anamnese e exame físico completo. O estudo oftalmológico abordou a medida da acuidade visual, a presença de distopia cantorum (telecanto), a análise da coloração da íris e o mapeamento de retina com retinografia colorida (Figura 3). A medida da distopia cantorum foi obtida através do cálculo do índice W, resultante de uma análise envolvendo a medida de três pontos anatômicos entre os dois olhos, conforme a figura 4. A distopia está presente quando o índice W resulta em valor maior ou igual a 1,95 mm(4).
Exames otológicos envolveram otoscopia, impedanciometria e audiometria computadorizada. Análise de imagens, incluindo fotografias do telecanto, hipopigmentação da íris e retina (retinografia colorida), além de alterações na pigmentação do cabelo e pele foram realizadas.
Família 2
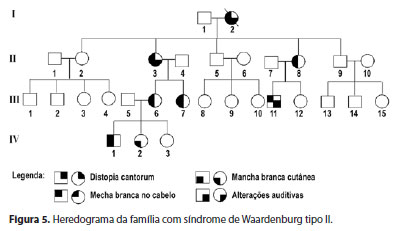
Paciente de 53 anos, feminino, procedente de Capitão Enéas, Minas Gerais, foi avaliada no mesmo Serviço da família 1, pois apresentava pterígio bilateral com crises de inflamação recorrente. A paciente apresentava uma mecha branca na região frontal, manchas acrômicas em membros inferiores e surdez moderada (Figura 1). Por meio de uma avaliação auditiva e uma audiometria, detectou-se a presença de surdez neurossensorial. Como a paciente não apresentava distopia cantorum, confirmou-se o diagnóstico de SW tipo II. Iniciou-se um estudo envolvendo toda a família da paciente, composta por 24 membros, nas últimas quatro gerações, para determinar o padrão de herança e a expressividade da doença (Figura 5).
Os pacientes afetados tinham idade entre 4 e 53 anos (média de 25,3 anos). A avaliação clínica incluiu anamnese e exame físico. O estudo oftalmológico abordou a medida da acuidade visual, presença de distopia cantorum (telecanto), análise da coloração da íris e mapeamento de retina. Exames otológicos envolveram otoscopia, impedanciometria e audiometria computadorizada. Análise de imagens, incluindo fotografias do telecanto e alterações na pigmentação do cabelo e pele foram realizadas.
O estudo foi submetido e aprovado pelo Comitê de Ética Institucional. Consentimento informado, livre e esclarecido foi obtido de cada paciente ou seu responsável legal, antes da inclusão no estudo.
RESULTADOS
Família 1
O exame clínico das últimas quatro gerações da família revelou que dos 15 membros (4 homens e 11 mulheres), 11 (73,33%) apresentaram características da SW. Desses 11 pacientes, a distribuição por sexo foi de 3 (27,27%) masculino e 8 (72,72%) do feminino. Não havia história de consanguinidade na família. O heredograma da família revelou um padrão de transmissão autossômico dominante com penetrância incompleta e expressividade variável (Figura 2). Dez membros apresentaram dystopia cantorum. Desses 10, 2 apresentaram surdez neurossensorial, 6 hipopigmentação da íris confirmada por uma significativa hipoplasia do epitélio pigmentado observado no exame de biomicroscopia e 6 topete branco, caracterizando o tipo I da SW por possuírem dois critérios maiores, segundo Read, Newton(4). Sinófris foi observado em 4 pacientes afetados pela síndrome. O probando foi submetido à cirurgia para implante coclear bilateral aos 2 anos e já responde a comandos verbais e fala com dicção compreensível.Todos os pacientes portadores de hipopigmentação da íris receberam óculos solares com filtro contra UV-A e UV-B, além da correção óptica, quando necessário.
Família 2
Nesta família, composta por 24 descendentes distribuídos em quatro gerações, características da SW estiveram presentes em 33,33% (6 homens e 2 mulheres) (Figura 1). A primeira geração foi considerada pelos relatos dos membros das gerações seguintes. Desses 8, a distribuição por sexo foi de 2 (25%) masculino e 6 (75%) feminino. Não havia história de consanguinidade na família. O heredograma da família revelou um padrão autossômico dominante (Figura 5). Nenhum membro apresentou distopia cantorum. Três pacientes apresentaram surdez neurossensorial associada a topete branco e manchas acrômicas confluentes pelo corpo, caracterizando o tipo II da SW por possuírem dois critérios maiores e ausência de distopia, segundo alguns autores(5). A probanda foi submetida a implante de prótese auditiva com recuperação dos limiares auditivos normais e à exérese do pterígio no olho esquerdo.
DISCUSSÃO
A SW é uma doença de distribuição universal, sem predileção para raça e gênero(4). A prevalência estimada do tipo I foi de 1:42.000 na população geral e 1:143 (1,43%) em portadores de surdez congênita(4). A SW apresenta um padrão de herança autossômica dominante com penetrância incompleta e expressividade variável, especialmente nos tipos I e II(9). A expressiva heterogeneidade clinica da SW é atribuída a uma rede complexa de interação entre seis genes identificados até o momento: o gene PAX3, principal responsável pela SW tipo I e III, os genes MITF, SOX10 e SNAI2 na SW tipo II e os genes EDN3 e EDNRB na SW tipo IV. Todos esses seis genes estão envolvidos dentro de uma rede de interações que determinam falhas na embriogênese das células derivadas da crista neural e caracterizam a diversidade fenotípica da SW(1).
No presente estudo, relatou-se duas famílias, ambas do norte do estado de Minas Gerais. A primeira apresentou o padrão clínico tipo I, e segunda família, tipo II. A distopia cantorum é a característica mais comum da SW tipo I, podendo ter uma incidência de 99% nesse subgrupo(4). Este deslocamento lateral do canto interno dos olhos se tornou o recurso diagnóstico mais confiável para classificação do tipo I, devido a sua elevada frequência(1). Já a hipertricose medial das sobrancelhas (sinófris) é mais frequente no tipo I (85%) do que no tipo II (25%)(5). A família com SW tipo I apresentou 78,57% dos seus membros com distopia cantorum e 28,57% deles com sinofris. A segunda família, formada por indivíduos com a SW tipo II, apresentou um padrão de transmissão autossômica dominante com penetrência incompleta e expressividade variável. Nenhum membro desta família apresentou distopia cantorum e hipopigmentação da íris.
A heterocromia de íris pode ser completa ou parcial(5). Na parcial, existe a presença de cores diferentes em um mesmo olho. Alguns autores encontraram heterocromia parcial em 4,2% dos indivíduos com SW tipo I e em 27,5% dos indivíduos com SW tipo II(5). Ohno et al., descreveram a presença de heterocromia de íris completa ou parcial em 30% dos indivíduos com SW tipo I e II(10). Iris extremamente azul bilateralmente pode fazer parte do quadro ocular em 10% de todos os pacientes com diagnóstico de SW e esta coloração é devido à intensa hipoplasia do seu epitélio pigmentado(1). No presente estudo, hipopigmentação da íris esteve presente em 42,85% na família com SW tipo I. Uma característica desta alteração é que a hipopigmentação da íris se apresentou simetricamente entre os dois olhos e no probando com SW tipo I foram observadas íris extremamente azuladas (Figura 1). Neste paciente foi observada uma significativa rarefação e mobilização do epitélio pigmentado da retina nos dois olhos, alteração descrita na literatura e que geralmente correspondente ao mesmo território alterado na íris(10).
A perda auditiva na SW é neurossensorial, de caráter congênito, não progressivo, podendo ser uni ou bilateral e variar em níveis de intensidade(4). Outros autores, encontraram uma penetrância de perda auditiva neurossensorial na SW tipo I de 69% e de 87% na SW tipo II(11). A hipoacusia decorre de uma degeneração cocleossacular e ausência completa de pigmentação na estria vascular da cóclea culminando em uma atrofia do órgão de Corti(3,12). No presente estudo a surdez neurossensorial esteve presente em 14,28% dos indivíduos com SW tipo I e em 12,50% na família com SW tipo II.
Em relação à mecha branca frontal, esta pode estar presente desde o nascimento ou surgir na primeira infância, como também pode desaparecer com a idade adulta(4). Pingault et al., relatam que a alteração da cor dos cabelos está presente em pelo menos um terço dos tipos I e II(1). No presente estudo o topete branco esteve presente em 50% na família com SW tipo I e 29,16% na SW tipo II.
A expressividade da SW varia amplamente na literatura, sendo apresentada de forma diferente em cada família acometida. Na avaliação das famílias apresentadas neste estudo foram encontradas características clínicas diferentes na forma de apresentação e nas suas prevalências. Por exemplo, a penetrância de distopia cantorum e hipopigmentação da íris foram expressivas na família com SW tipo I (78,57 e 50%, respectivamente), mas a perda auditiva foi limitada (14,28%) e observada apenas em dois indivíduos (III-6 e IV-7) dos 14 membros da família. Na família com SW tipo II, não foram observadas alterações oftalmológicas, apenas surdez e manchas hipocrômicas no cabelo e na pele.
A presença de alterações oftalmológicas na SW tipo I exige uma abordagem oftalmológica direcionada a todos os membros da família baseada na prevenção de doenças oculares e na reabilitação visual destes pacientes com deficiência de pigmentação em dois tecidos de suma importância na fisiologia visual, a íris e a retina. Todos os pacientes foram submetidos a exame refracional e receberam a prescrição da correção. Nos casos de emetropia, foram indicados óculos solares com filtros contra os raios ultravioletas.
CONCLUSÃO
O presente estudo mostra a importância do oftalmologista no auxílio do diagnóstico deste raro quadro sistêmico, uma vez que inclui alterações oftalmológicas como distopia cantorum, hipopigmentação da íris e da retina. A distopia cantorum é o principal critério diagnóstico para diferenciar o tipo I do II e deve ser feita por oftalmologista treinado. Aconselhamento genético e cuidados para proteger um olho com deficiência de pigmentação na íris e na retina foram oferecidos aos pacientes. Investigações envolvendo amostras maiores, com enfoque em bases genéticas e moleculares são necessárias para elucidar as consequências destas alterações clínicas e melhorar nossa compreensão sobre os mecanismos envolvidos nesta patologia, a fim de podermos oferecer um aconselhamento genético criterioso e um tratamento mais completo aos pacientes.
REFERÊNCIAS
1. Pingault V, Ente D, Dastoto-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Human Mutat. 2010;31(4):391-406.
2. Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrowns and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet. 1951;3(3):195-253.
3. Nakashima S, Sando I,Takahashi H, Hashida Y. Temporal bone histopathologic findings of the Waardenburg's syndrome: a case report. Laryngoscope. 1992;102(5):563-67.
4. Read AP, Newton VE. Waardenburg syndrome. J Med Genet. 1997;34(8):656-65.
5. Liu XZ, Newton VE, Read AP. Waardenburg syndrome type 2: phenotypic findings and diagnostic criteria. Am J Med Genet. 1995;55(1):95-100.
6. Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH Jr, Beithton P, et al. Waardenburg syndrome (WS) tipe 1 is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: first report of the WS Consortium. Am J Hum Genet. 1992;50(5):902-13.
7. Hageman MJ, Delleman JW. Heterogeneity in Waardenburg syndrome. Am J Hum Genet. 1977;29(5):468-85.
8. Fraser Gr. The cause of profound deafness in childhood. Baltimore: John Hopkins University Press; 1976
9. Arias S. Genetic heterogeneity in the Waardenburg syndrome. Birth Defects Orig Artic Ser. 1971;7(4):87-101.
10. Ohno N, Kiyosawa M, Wang WF, Takase H, Mochizuki M. Clinical findings in Japanese patients with Waardenburg syndrome type 2. Jpn J Ophthalmol. 2003;47(1):77-84.
11. Newton VE, Read AP. Waardenburg Syndrome. Audiol Med. 2003;1(1):77-88.
12. Steel KP, Barkway C. Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development. 1989;107(3):453-63.
13. Partington MW. Waardenburg's syndrome and heterochromia iridum in a deaf school population. Can Med Assoc J. 1964;90:1008-17.
 Endereço para correspondência:
Endereço para correspondência:
Luciano Sólia Násser
Rua Walter F. Barreto, 57
Montes Claros (MG) 39401-347 - Brasil
E-mail: [email protected]
Submetido para publicação: 26 de julho de 2012
Aceito para publicação: 6 de outubro de 2012
Financiamento: Não houve financiamento para este trabalho.
Divulgação de potenciais conflitos de interesse: L.S.Nasser, Nenhum; L.M.R. Paranaíba, Nenhum; A.C.Frota, Nenhum; A.Gomes, Nenhum; G.Versiani, Nenhum; H.Martelli.Jr., Nenhum.
Comitê de Ética: Unimontes 2781/2011.
Trabalho realizado na Universidade Estadual de Montes Claros, Unimontes, Montes Claros, Minas Gerais, Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.