Arq. Bras. Oftalmol. 2001; 64 (5): 10.1590/S0004-27492001000500020
Total: 2892
Ana Tereza Ramos Moreira1; Hilton Iran Ruthes1; Silvane Bigolin2
DOI: 10.1590/S0004-27492001000500020
RESUMO
A miastenia gravis é uma doença crônica, caracterizada por fatigabilidade anormal de músculos estriados, podendo acometer grupos musculares isolados ou tornar-se generalizada. Os autores descrevem um caso de miastenia gravis congênita generalizada e oftalmoplegia parcial em um paciente de 10 anos de idade, portador de sintomas sistêmicos motores e de ausência na adução, abdução e elevação em ambos os olhos e com ptose palpebral bilateral, sendo reduzida à função de infraversão. O paciente foi diagnosticado aos dois anos e seis meses, sendo iniciado tratamento com piridostigmina em doses subterapêuticas, sem alteração importante no quadro clínico. Aos quatro anos de idade, procurou esta instituição, sendo ajustada à dose da medicação, seguindo-se melhora significativa dos sintomas motores sistêmicos, melhora parcial da ptose palpebral e sem alteração na oftalmoplegia externa.
Descritores: Miastenia gravis; Miastenia gravis; Piridostigmina/brometo; Oftalmoplegia; Blefaroptose; Relato de Caso
ABSTRACT
Myasthenia gravis is a chronic disease characterized by abnormal fatigability of striated muscles which may involve isolated muscle groups or become generalized. The authors report a case of generalized congenital myasthenia gravis and partial ophthalmoplegia involving a 10-year-old boy with systemic symptoms of absence of adduction, abduction and elevation in both eyes and with bilateral palpebral ptosis, and reduced infraversion function. The patient was diagnosed at two years and six months of age, when treatment with pyridostigmine was started at subtherapeutic doses, with no significant changes in clinical signs and symptoms. At four years of age, the patient was taken to this institution and the dose of his medication was adjusted, with significant improvement of systemic motor systems, partial improvement of palpebral ptosis, and no change in external ophthalmoplegia.
Keywords: Myasthenia gravis; Myasthenia gravis; Pyridostigmine bromide; Ophthalmoplegia; Blepharoptosis; Case report
INTRODUÇÃO
A miastenia gravis é uma doença crônica, caracterizada por fatigabilidade anormal de músculos estriados, podendo acometer grupos musculares isolados ou tornar-se generalizada(1).
É uma condição rara em crianças (cerca de 10% dos casos ocorrem antes da puberdade), nas quais o diagnóstico pode ser difícil devido às apresentações atípicas(2). Os casos de miastenia gravis em crianças podem ser divididos em neonatal, congênitos e juvenis, cada um apresentando uma entidade clínica distinta e cujo diagnóstico deve ser correto, diferenciando entre os subtipos, visto ser a terapêutica escolhida de acordo com a forma envolvida(3).
A causa da miastenia gravis é um defeito, autoimune ou não, no mecanismo da acetilcolina na junção neuromuscular. Muitos casos estão associados à hiperplasia ou tumor do timo. Os músculos extra-oculares são particularmente sensíveis às alterações da junção neuromuscular que ocorrem na miastenia gravis, as quais podem provocar, oftalmoplegia e ptose palpebral, poupando a pupila e a acomodação(1).
As síndromes miastênicas congênitas são desordens incomuns, mas desafiantes, podendo ocasionar morte ao nascimento ou, em casos leves, não ser diagnosticadas até o paciente ser exposto a agentes bloqueadores neuromusculares. Casos de miastenia gravis congênita têm sido relatados desde 1949 e é descrita incidência familiar, embora não se conheça o padrão genético envolvido(4-5).
Apresentamos o caso de uma criança de 10 anos e 11 meses com miastenia gravis congênita generalizada e oftalmoplegia externa, em acompanhamento em nosso serviço.
RELATO DO CASO
G.A.L., masculino, branco, 10 anos e 11 meses, foi encaminhado ao Serviço de Oftalmologia do Hospital de Clínicas da Universidade Federal do Paraná (UFPR) pelo Serviço de Neuromuscular da mesma instituição, por apresentar limitação da motilidade ocular em ambos os olhos e ptose palpebral bilateral. A criança nasceu no município de Barbosa Ferraz-PR, de gestação a termo, em regulares condições. Apresentava choro fraco, movimentação débil, dificuldade de sucção com engasgos freqüentes, ptose palpebral bilateral e oftalmoplegia. Persistiu com o quadro, somado a hipotonia sistêmica importante e dificuldade de deambulação. Apresentou diversas internações hospitalares antes dos dois anos de idade por amigdalites de repetição. Aos dois anos e seis meses, a família procurou atendimento médico no interior do estado de São Paulo, ocasião em que foi diagnosticada miastenia gravis e foi iniciado o tratamento com inibidor da acetilcolinesterase (piridostigmina), sem o correto ajuste da droga de acordo com o crescimento da criança. Aos quatro anos de idade, o menor foi encaminhado ao Hospital de Clínicas da UFPR, onde teve a dose da medicação reajustada, levando a melhora importante dos sintomas sistêmicos e melhora parcial da ptose palpebral, persistindo, contudo a oftalmoplegia externa.
Os pais da criança são saudáveis e primos em primeiro grau, sendo também os avós paternos primos em primeiro grau. Há relato na história familiar de avó materna com diminuição da força muscular nos membros superiores e inferiores e de casos de estrabismo (avó paterna, tia paterna, primo materno).
A criança foi submetida à biópsia muscular e teste de estímulo repetitivo aos cinco anos de idade, sendo ambos sugestivos de miastenia gravis congênita.
Atualmente, a criança encontra-se em uso de brometo de piridostigmina na dose de 60 mg de seis em seis horas.
Ao exame oftalmológico em uso da medicação, o paciente apresentava-se com paralelismo ocular em posição primária da mirada, motilidade ocular intrínseca preservada, ausência de adução, abdução e elevação em ambos os olhos, redução na infraversão e ptose palpebral bilateral leve (Figura). Apresentava acuidade visual sem correção de 20/25 em ambos os olho. Biomicroscopia e fundoscopia normais. Ao exame de estereopsia realizado através do teste de Titmus apresentava 50 segundos de arco. A única função muscular externa encontrada foi a dos músculos retos inferiores e mesmo assim com redução da função de aproximadamente -2 (Figura).

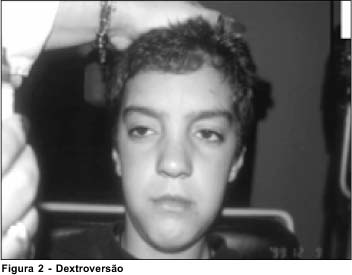

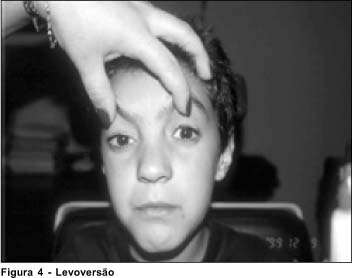
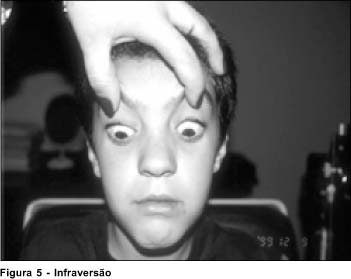
DISCUSSÃO
A miastenia gravis possui prevalência de dois a quatro para cada 100.000 habitantes e é ainda menos comum em crianças(2,5). Os casos em crianças e adolescentes respondem por 11 a 24% de todos os pacientes com miastenia gravis e podem ser divididos em neonatal, congênita e juvenil(3,6).
A miastenia gravis neonatal é uma desordem transitória, que pode afetar crianças nascidas de mães com miastenia gravis, e é causada pela passagem transplacentária de anticorpos IgG contra o receptor de acetilcolina. Na miastenia gravis juvenil, o distúrbio inicia após os seis meses de vida, apresenta-se similar à forma adulta e caracteriza-se por anticorpos contra o receptor de acetilcolina. Já a forma congênita é desprovida de anticorpos contra os receptores e as mães são normais(3). Millichap, em uma análise de 447 pacientes com miastenia gravis, encontrou 1% dos casos com miastenia congênita, 2% com neonatal e 8% com a forma juvenil, com as primeiras manifestações após o primeiro ano de vida(7).
A miastenia gravis congênita é um grupo heterogêneo de desordens nas quais vários componentes da junção neuromuscular são disfuncionais. Não existe nenhuma evidência histopatológica ou sorológica de processo imune contra o receptor de acetilcolina, assim como não há sinais de doença na mãe(3). Os sintomas manifestam-se tipicamente ao nascimento ou nos primeiros anos de vida e constituem-se de dificuldades para a alimentação, sucção e deglutição, hipotonia, fraqueza muscular, ptose palpebral, oftalmoplegia externa, diminuição da expressão facial e envolvimento principalmente dos grupos musculares cervicais e do dorso, podendo as manifestações oculares ser as primeiras a se manifestar(8-9). Em alguns casos, existe severo acometimento extra-ocular na infância com fraqueza generalizada leve, a qual pode desenvolver-se concomitantemente ou anos mais tarde(10).
A raridade da doença pode fazer com que os sintomas sejam erroneamente interpretados pelo pediatra, atrasando o diagnóstico e podendo colocar em risco a vida da criança. Schlezinger e Millichap relatam em seus estudos que o óbito pode ocorrer logo após o nascimento, se não houver o pronto reconhecimento da miastenia gravis(7,11).
As síndromes miastênicas congênitas podem ser devidas a alterações pré-sinápticas, sinápticas ou pós-sinápticas da junção neuromuscular. As mutações no receptor de acetilcolina, que aumentam ou diminuem a resposta à acetilcolina, são causa comum de miastenia gravis congênita pós-sináptica. Também são descritas ausência da forma assimétrica de acetilcolinesterase na lâmina basal sináptica e deficiência de receptores de acetilcolina(8-10).
O diagnóstico da miastenia gravis congênita baseia-se nas manifestações clínicas, teste do estímulo repetitivo, resposta à terapia anticolinesterásica e ausência de anticorpos contra o receptor de acetilcolina. Podem também ser utilizados testes farmacológicos, especialmente na caracterização da forma ocular. Em crianças maiores e adultos, administra-se lentamente 2 mg de cloreto de edrofônio por via endovenosa, seguido de 8mg em bolo, o que produz redução da ptose. Em crianças com menos de 10 Kg, a dose deve ser reduzida ou substituída por neostigmina intramuscular 20 minutos após injeção endovenosa de atropina(1,11-12).
O tratamento da miastenia gravis congênita é suportivo e os inibidores da acetilcolinesterase podem ser úteis em algumas formas. Timectomia, imunossupressão e plasmaferese não são efetivos e a remissão espontânea não é esperada(8). Em adultos, um novo método relativamente sensível para o diagnóstico da ptose miastênica, foi relatado. É o teste do gelo ou "Ice test", que consiste na mensuração da fenda palpebral antes e após aplicação de gelo sobre a pálpebra ptótica. Porém a sensibilidade desse teste decresce consideravelmente em pacientes com ptose completa(13). A manifestação ocular inicial, na maioria dos casos, envolve mais de um músculo extra-ocular, com queixas comuns de ptose palpebral e diplopia. O envolvimento extra-ocular pode ser tão severo a ponto de causar oftalmoplegia externa completa(11). É descrito na literatura que, embora a terapêutica possa resultar em melhora significativa dos sintomas sistêmicos, a resposta ocular geralmente é pobre, sem grandes alterações no curso natural da doença(5,7,11). Jones descreve a existência de diferenças antigênicas nos receptores de acetilcolina dos músculos extra-oculares comparados aos músculos esqueléticos e sugere que tais diferenças poderiam explicar o envolvimento ocular e a refratariedade ocular ao tratamento(12). Alguns autores têm sugerido o uso de imunossupressores (corticóides) para a melhora da oftalmoplegia, mas com resultados não comprovados no grupo da miastenia gravis congênita(14).
O caso apresentado tem a peculiaridade de apresentar paralisia dos músculos reto lateral, reto medial, reto superior, oblíquo superior e oblíquo inferior em ambos os olhos, poupando parcialmente o músculo reto inferior e sendo mantido o paralelismo na posição primária da mirada. A criança apresenta também sintomas motores sistêmicos importantes e ptose palpebral bilateral, sendo que o tratamento com piridostigmina em doses adequadas resulta em melhora importante dos sintomas sistêmicos e melhora parcial da ptose.
É importante salientar que em diversas ocasiões antes do diagnóstico a criança esteve sob cuidados médicos em ambiente hospitalar, por apresentar episódios de amigdalite de repetição e mesmo com acompanhamento freqüente pelo pediatra, o diagnóstico de miastenia gravis foi tardio. Somando-se ao diagnóstico tardio, a terapêutica inicial estabelecida foi com doses inapropriadamente baixas, ambos os fatos contribuindo para que a criança ficasse exposta a um alto risco de complicações, principalmente respiratórias e potencialmente fatais. É preciso que se estabeleça em todos os pacientes com miastenia gravis o diagnóstico precoce e correto e o ajuste das doses de medicação de acordo com o crescimento da criança, sob pena de risco à vida do paciente.
REFERÊNCIAS
1. Taylor D. Paediatric Ophthalmology. 2nd ed. Oxford: Blackwell Scientific; 1997.
2. Vial C, Charles N, Chauplannaz G, Bady B. Myasthenia gravis in childhood and infancy. Usefulness of electrophysiologic studies. Arch Neurol 1991;48: 847-9.
3. Andrews I, Massey JM, Sanders DB. Acetylcoline receptor antibodies in juvenile myasthenia gravis. Neurology 1993;43:977-82.
4. Engel AG. Congenital myasthenia syndromes. Neurol Clin 1994;12:401-37.
5. Bundey S. A genetic study of infantile and juvenile myasthenia gravis. J Neurol Neurosurg Psychiatry 1972;35:41-51.
6. Snead OC, Benton JW, Dwyer D, Morley BJ, Kemp GE, Bradley RJ, et al. Juvenile myasthenia gravis. Neurology 1980;30(Pt 1):732-9.
7. Millichap JG, Dodge PR. Diagnosis and treatment of myasthenia gravis in infancy, childhood, and adolescence. A study of 51 patients. Neurology 1960; 10:1007-14.
8. Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes - recent advances. Arch Neurol 1999;56:163-7.
9. Oosterhuis HJ. The natural course of myasthenia gravis: a long term follow up study. J Neurol Neurosurg Psychiatry 1989;52:1121-7.
10. Gordon N. Congenital myasthenia. Dev Med Child Neurol 1986;28:810-3.
11. Schlezinger NS, Fairfax WA. Evaluation of ocular signs and symptoms in myasthenia gravis. Arch Ophthalmol 1959;62:985-90.
12. Jones KJ, North KN. External ophthalmoplegia in neuromuscular disorders: case report and review of the literature. Neuromuscul Disord 1997;7:143-51.
13. Golnik KC, Pena R, Lee AG, Eggenberger ER. An ice test for the diagnosis of myasthenia gravis. Ophthalmology 1999, 106: 1282-6.
14. Kupersmith MJ, Moster M, Bhuiyan S, Warren F, Weinberg H. Beneficial effects of corticosteroids on ocular myasthenia gravis. Arch Neurol 1996; 53:802-4.
1Chefe do Serviço de Oftalmologia, Hospital de Clínicas da Universidade Federal do Paraná.
2 Residente de Oftalmologia, Hospital de Clínicas da Universidade Federal do Paraná.
3 Acadêmica de medicina, Universidade Federal do Paraná.
Endereço para correspondência: R. Ubaldino do Amaral 1062 / ap. 202 - Curitiba (PR) CEP 80060-190. E-mail: [email protected]
Nota Editorial: Pela análise deste trabalho e por sua anuência sobre a divulgação desta nota, agradecemos ao Dr. Carlos Ramos Souza-Dias.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.