INTRODUCTION
The cornea is the primary refractive element of the visual system. It is principally composed of stroma, which accounts for approximately 90% of its thickness(1). The corneal stroma is covered anteriorly by four to six layers of stratified, nonkeratinized epithelium; posteriorly, it is covered by a single endothelial layer, which is in direct contact with the aqueous humor(1-3). The corneal stroma consists of collagen fibers, extracellular matrix (ECM), and keratocytes, which exhibit dendritic morphology and function in the synthesis of collagen fibrils. The uniform arrangement of these collagen fibers allows corneal transparency(4,5).
A number of diseases have been shown to affect the arrangement and resistance of corneal collagen fibers. Keratoconus is defined as a noninflammatory disease, generally involving bilateral corneal ectasia, characterized by stromal thinning and loss of rigidity, leading to irregular asymmetric astigmatism and poor vision. In the majority of cases, the refractive error induced by keratoconus can be corrected with the use of rigid contact lenses. Nevertheless, a proportion of cases in which progression of the corneal irregularity results in significant visual deterioration require more invasive therapeutic options, such as intracorneal implants or corneal transplantation(6).
Recently, Seiler et al. proposed the use of crosslinking (CXL) for the treatment of keratoconus. This treatment is based on the chemical induction of interfibrillar and intrafibrillar covalent bonds in the corneal stroma, thus strengthening the cornea. CXL relies on photosensitive oxidation induced by the combination of riboflavin (vitamin B2) and ultraviolet A (UVA) light, thereby increasing the biomechanical resistance and stability of the cornea(7,8). Other indications for crosslinking have been reported, including the treatment of post-refractive surgery ectasia, bullous keratopathy, ocular infections, and corneal necrosis, among others(8).
The objective of the present study was to culture quiescent human keratocytes and evaluate the effect of UVA light and riboflavin on human corneal keratocytes in vitro.
METHODS
Study design
In all the groups assigned to receive 0.1% riboflavin (10 mg riboflavin-5-phosphate dissolved in 10 mL of 20% dextran T-500 [400 mOsm/mL]; Ophtalmos®, São Paulo, SP Brazil), a layer of riboflavin was applied to the surface of the plate, covering all cells for 30 minutes. This procedure was performed before continuous exposure to UVA light (IROC®, Germany) at 370 nm with a surface irradiance of 3 mW/cm2, with a total radiant exposure of 5.375 J/cm2 in cases in which riboflavin and UVA light were combined. The riboflavin concentration and UV wavelength used in the present study were identical to those used clinically. The twelve experimental groups are described below:
Keratocyte culture in the presence of 500 µL of collagen at 0.1 g/mL
8. Cells were exposed to 0.1% riboflavin for 30 minutes;
9. Cells were exposed to UVA light at 370 nm for 30 minutes;
10. Cells were simultaneously exposed to riboflavin and to UVA light for 30 minutes;
1. Cells were exposed to collagen alone;
2. Control group: keratocytes were not submitted to any of the abovementioned experimental conditions.
Keratocyte culture
Keratocytes were isolated from remaining corneoscleral ring donor corneas previously used in corneal transplant surgeries at the Department of Ophthalmology and Visual Sciences, Escola Paulista de Medicina (EPM), Universidade Federal de São Paulo (UNIFESP). Explants of corneal stroma were prepared in a laminar flow hood under sterile conditions. Explants were cultured in Dulbecco's modified eagle medium and Ham's nutrient mixture F12 (DMEM/F12-1:1) supplemented with 2% fetal bovine serum (FBS; Invitrogen, Gibco, Portland, OR, USA) in 24-well plates. Cells were cultured in a humidified incubator at 37°C in 5% CO2. Culture medium was changed three times a week for 3 weeks.
Cytotoxicity (MTT)
Twenty-four hours after treatment, RPMI medium (Invitrogen, Gibco, Portland, OR, USA) with 10% MTT solution was added and cells were incubated for 3 hours at 37°C. To solubilize formazan crystals, 100 µL of dimethyl sulfoxide (DMSO; Sigma Aldrich, St. Louis, MO, USA) was added to each well. Results were read using a spectrophotometer at a wavelength of 340 nm in an ELISA EXL800 plate reader (Universal Microplate Reader, Bio-Tek Instruments, Inc.). The cytotoxicity of each treatment was expressed by comparing absorption values with control values.
Apoptosis and necrosis (Hoechst 33342 and propidium iodide)
Cytomorphological assays to detect apoptotic and necrotic cells using Hoechst 33342 (Sigma Aldrich, St. Louis, MO, USA) and propidium iodide staining were performed following treatment in all groups. Cells were collected and centrifuged for 5 minutes at 1800 rpm. Next, a mixture of 3.5 µL of suspended cells, 1.5 µL of Hoescht 33342 (10 mg/mL), and 1.0 µL of propidium iodide was placed between a slide and coverslip. The reaction mixture was incubated and cells were observed using a fluorescence microscope with absorption spectra of 360 and 538 nm.
Immunofluorescence
Cells were cultured in 24-well culture plates (TPP, Zurich, Switzerland) on coverslips. After reaching 70% confluence, cells were divided into two groups: untreated, with immunofluorescence used only for cell characterization; treated, with immunofluorescence applied before and after CXL treatment to observe changes in protein expression.
Cells were incubated overnight with the following primary antibodies: lumican (keratocyte marker), Thy-1 (fibroblast marker), α-SMA (myofibroblast marker), and CK3 (epithelium marker, cell-characterization step only). Cells were then incubated with secondary anti-mouse antibody conjugated with fluorescein isothiocyanate (FITC). Slides were analyzed using a fluorescence microscope.
Statistical analysis
The software program used for the statistical analysis and for producing the graphs was GraphPad Prism, version 5.0 for Windows. The continuous, semi-continuous, and semi-categorical data were initially analyzed using the Kolmogorov-Smirnov test to determine how closely these data fitted to a normal (Gaussian) curve. The parametric data were represented as means and standard deviations of the means, and compared using Student's t-test, with or without Welch's correction, depending on the result of Levene's test used to determine the equality of variances.
For the parametric variables in independent samples, one-way analysis of variance (ANOVA) was used, followed by Dunnett's post-hoc test. The nonparametric data were represented as medians of quartiles and, when more than two samples were compared, the Kruskal-Wallis test was used followed by the Müller-Dunn post-test. Spearman's correlation coefficient was used to measure nonparametric correlations and Pearson's correlation was applied for parametric measurements.
An alpha (probability of making a type I error) ≤5% and a beta (probability of making a type II error) ≤20% were defined for the entire study. Probabilities <5% were considered statistically significant. For repeated measures, the Bonferroni method was used to correct cumulative type I errors.
RESULTS
Keratocyte culture
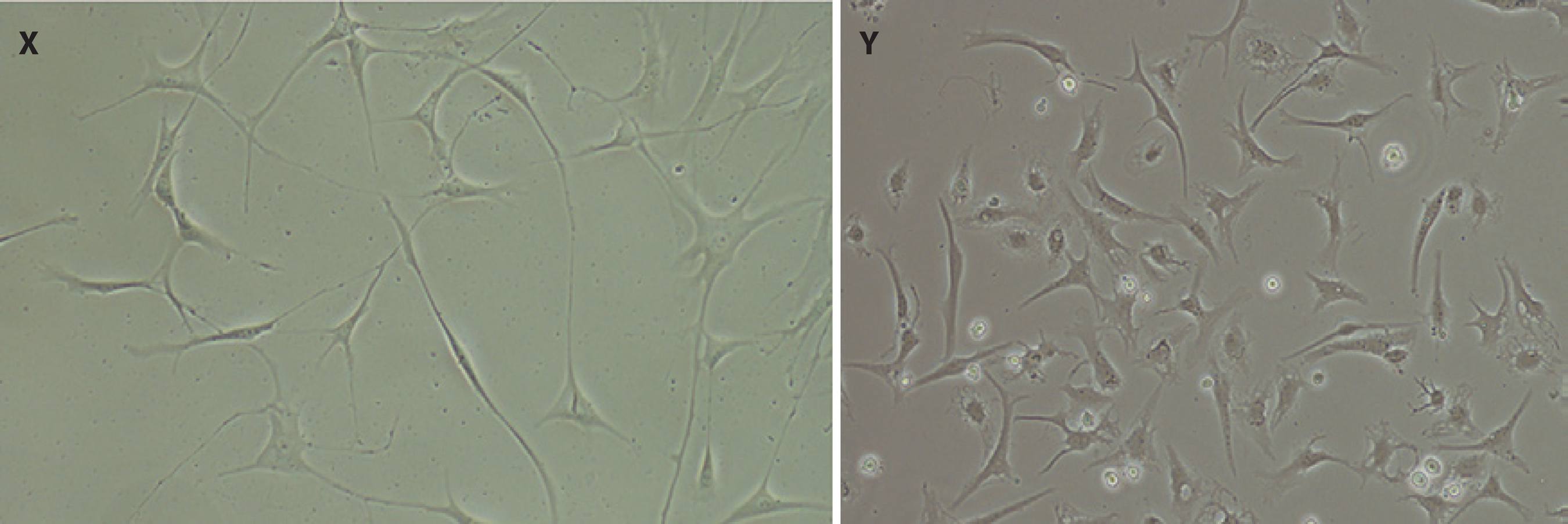
Corneal stromal cells were observed to have dendritic morphology after approximately 3 weeks' culture in DMEM/F12 medium containing 2% FBS, indicating cells remained quiescent during culture (Figure 1).
Cytotoxicity (MTT)
Cytotoxicity evaluations using MTT colorimetric assays 24 hours after treatment demonstrated a significantly lower number of metabolically active cells in cell cultures without collagen during exposure to UVA light, indicating poorer cell viability. No statistically significant differences were observed in the other treated groups compared to the control group (Figure 2).
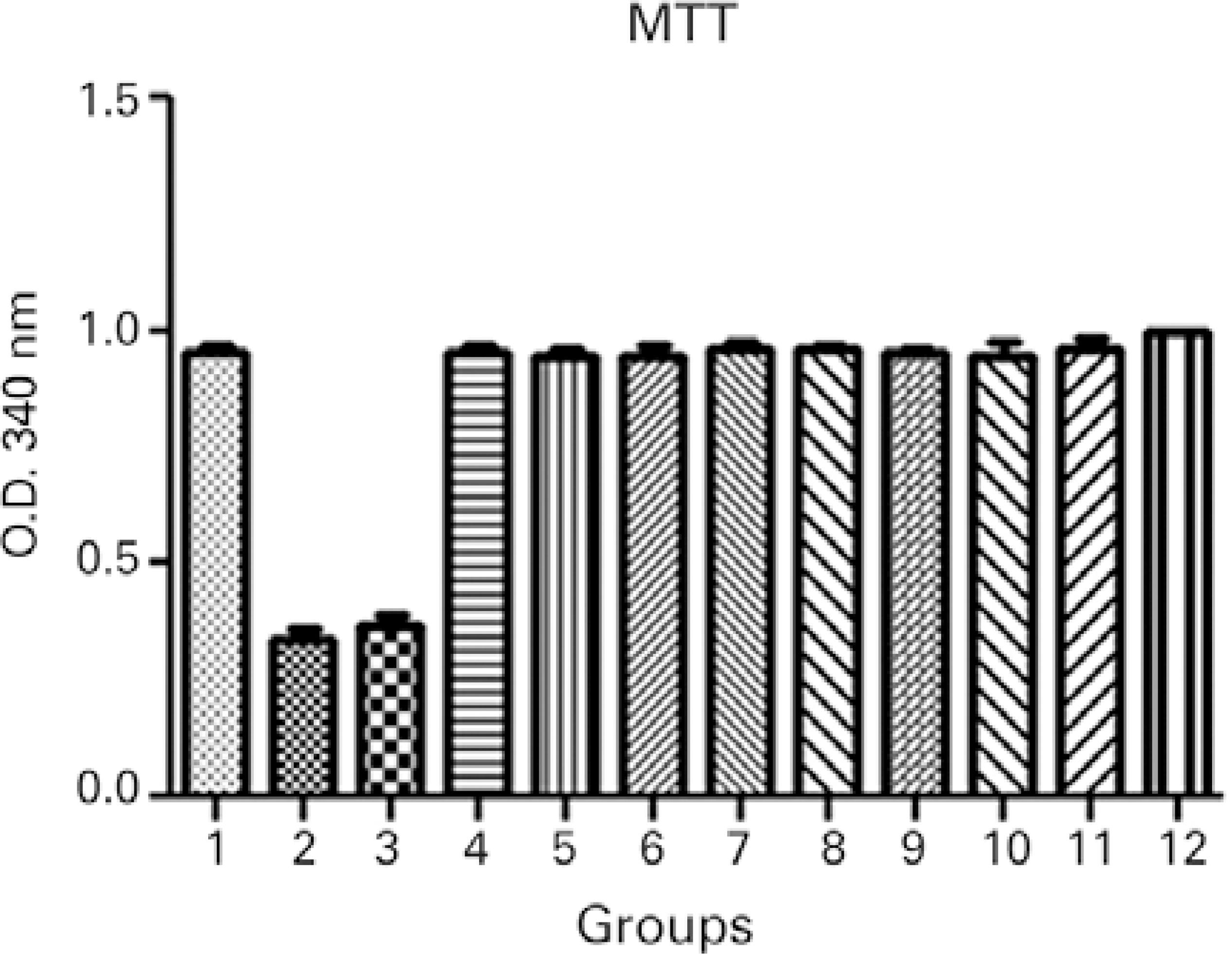
Figure 2 Cytotoxicity evaluations using MTT colorimetric assays 24 hours after treatment: 1, keratocyte culture + riboflavin; 2, keratocyte culture + UVA light; 3, keratocyte culture + riboflavin + UVA light; 4, keratocyte culture + 200 µL of collagen + riboflavin; 5, keratocyte culture + 200 µL of collagen + UVA light; 6, keratocyte culture + 200 µL of collagen + riboflavin + UVA light; 7, keratocyte culture + 200 µL of collagen; 8, keratocyte culture + 500 µL of collagen + riboflavin; 9, keratocyte culture + 500 µL of collagen + UVA light; 10, keratocyte culture + 500 µL of collagen + riboflavin + UVA light; 11, keratocyte culture + 500 µL of collagen; 12, control group. All groups were treated for 30 minutes.ANOVA= P<0.0001, Groups 2 and 3 versus Group 12 (control). Dunnett’s post-hoc test= P<0.001, Groups 2 and 3 versus Group 12 (control).
Apoptosis versus necrosis (Hoechst 33342 and propidium iodide)
To confirm the results of the MTT assay, cell viability of all treatment groups was evaluated using Hoechst 33342 and propidium iodide staining, which permitted the identification and quantification of viable cells, apoptotic cells (bright blue-stained cells), and necrotic cells (red-stained cells; Figure 3). Differential staining demonstrated a significantly greater number of necrotic and apoptotic cells in the groups without collagen during exposure to UVA light compared to the other treatment and control groups (Figures 4, 5, and 6).
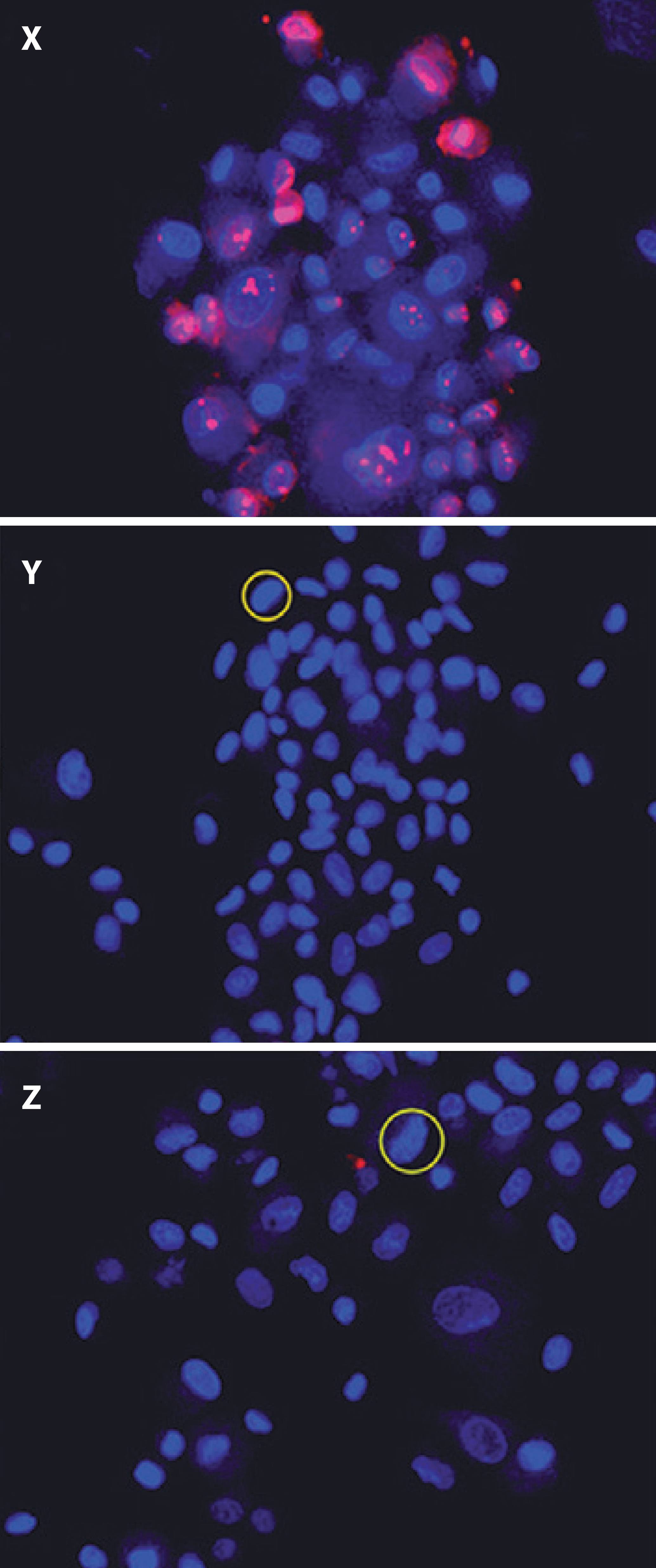
Figure 3 Cell viability in treatment groups evaluated using Hoechst 33342 and propidium iodide staining. X) Keratocyte culture + riboflavin + UVA light, without collagen; cells in necrosis are stained red. Y) Keratocyte culture + riboflavin + UVA light + 200 µL of collagen. Z) Keratocyte culture + riboflavin + UVA light + 500 µL of collagen. In Y and Z, chromatin was observed as highly compacted. Cells undergoing apoptosis are stained bright blue.
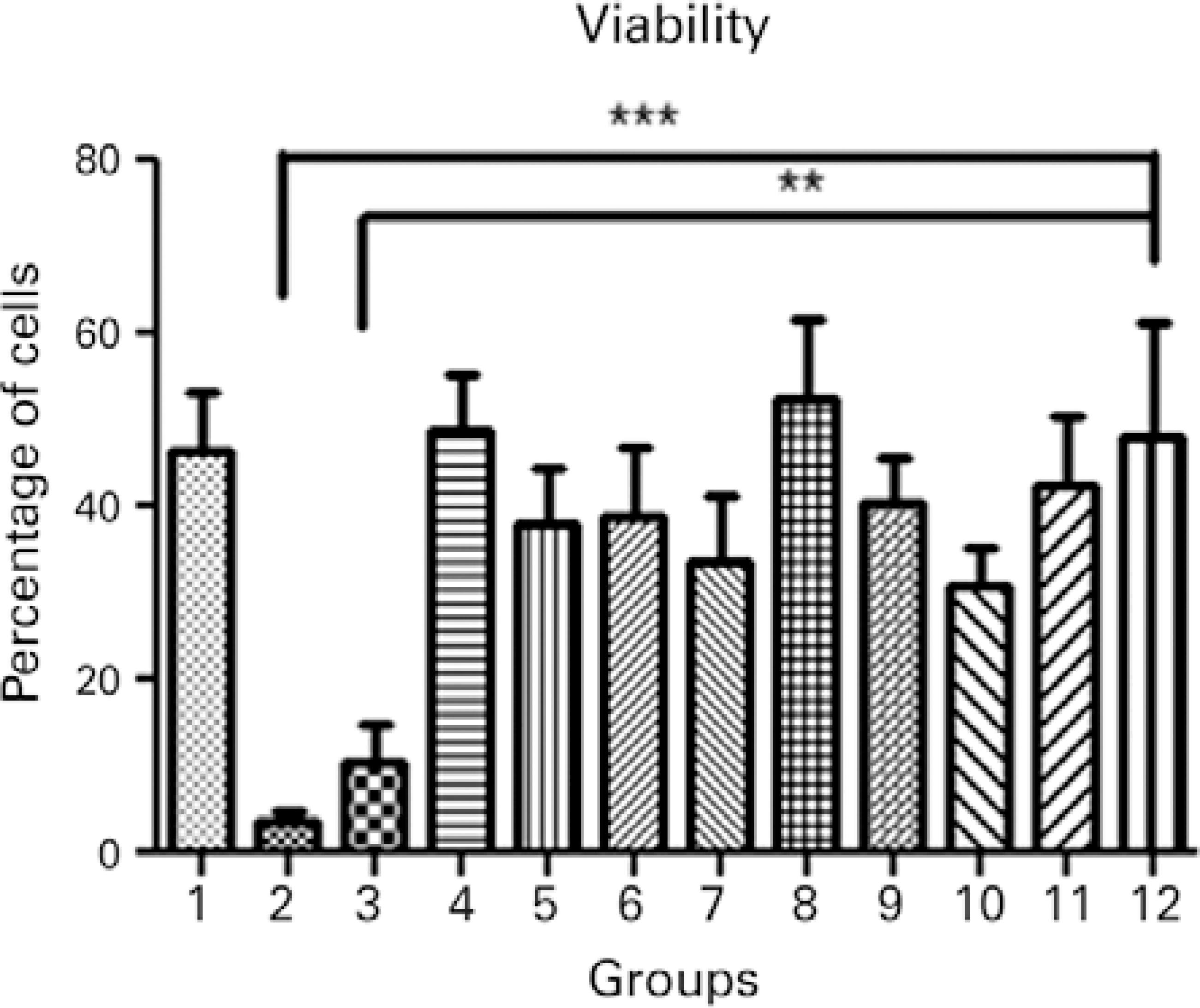
Figure 4 Cell viability in the different treatment groups: 1, keratocyte culture + riboflavin; 2, keratocyte culture + UVA light; 3, keratocyte culture + riboflavin + UVA light; 4, keratocyte culture + 200 µL of collagen + riboflavin; 5, keratocyte culture + 200 µL of collagen + UVA light; 6, keratocyte culture + 200 µL of collagen + riboflavin + UVA light; 7, keratocyte culture + 200 µL of collagen; 8, keratocyte culture + 500 µL of collagen + riboflavin; 9, keratocyte culture + 500 µL of collagen + UVA light; 10, keratocyte culture + 500 µL of collagen + riboflavin + UVA light; 11, keratocyte culture + 500 µL of collagen; 12, control group. All groups were treated for 30 minutes.ANOVA= p=0.0002; Group 2 versus Group 12 (control). Dunnett’s post-hoc test= p<0.001; Group 3 versus Group 12 (control); Dunnett’s post-hoc test; p<0.01.
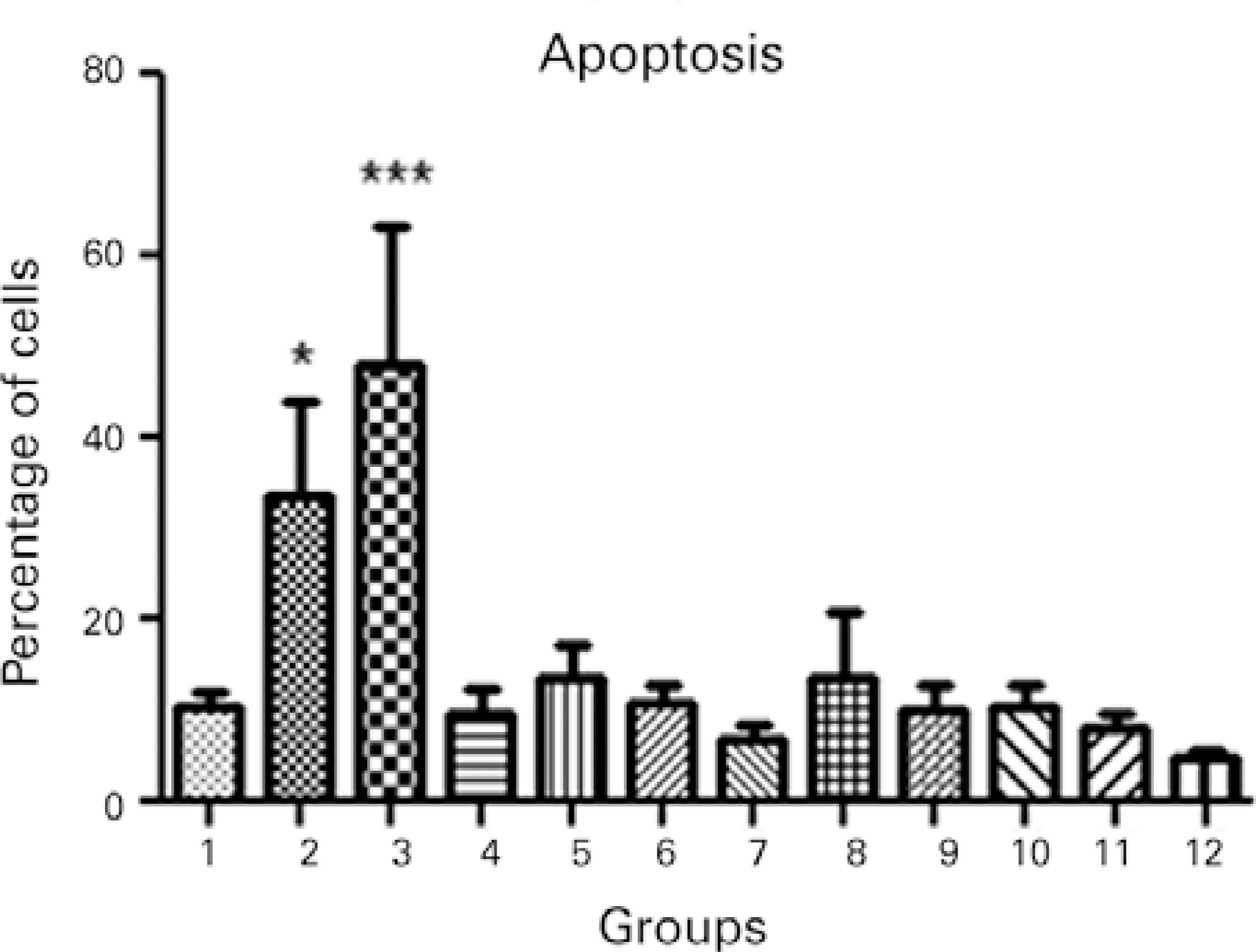
Figure 5 Apoptosis in the different treatment groups: 1, keratocyte culture + riboflavin; 2, keratocyte culture + UVA light; 3, keratocyte culture + riboflavin + UVA light; 4, keratocyte culture + 200 µL of collagen + riboflavin; 5, keratocyte culture + 200 µL of collagen + UVA light; 6, keratocyte culture + 200 µL of collagen + riboflavin + UVA light; 7, keratocyte culture + 200 µL of collagen; 8, keratocyte culture + 500 µL of collagen + riboflavin; 9, keratocyte culture + 500 µL of collagen + UVA light; 10, keratocyte culture + 500 µL of collagen + riboflavin + UVA light; 11, keratocyte culture + 500 µL of collagen; 12, control group. All groups were treated for 30 minutes.ANOVA= P<0.0001, Group 2 versus Group 12 (control). Dunnett’s post-hoc test= P<0.05, Group 3 versus Group 12 (control); Dunnett’s post-hoc test, P<0.001.
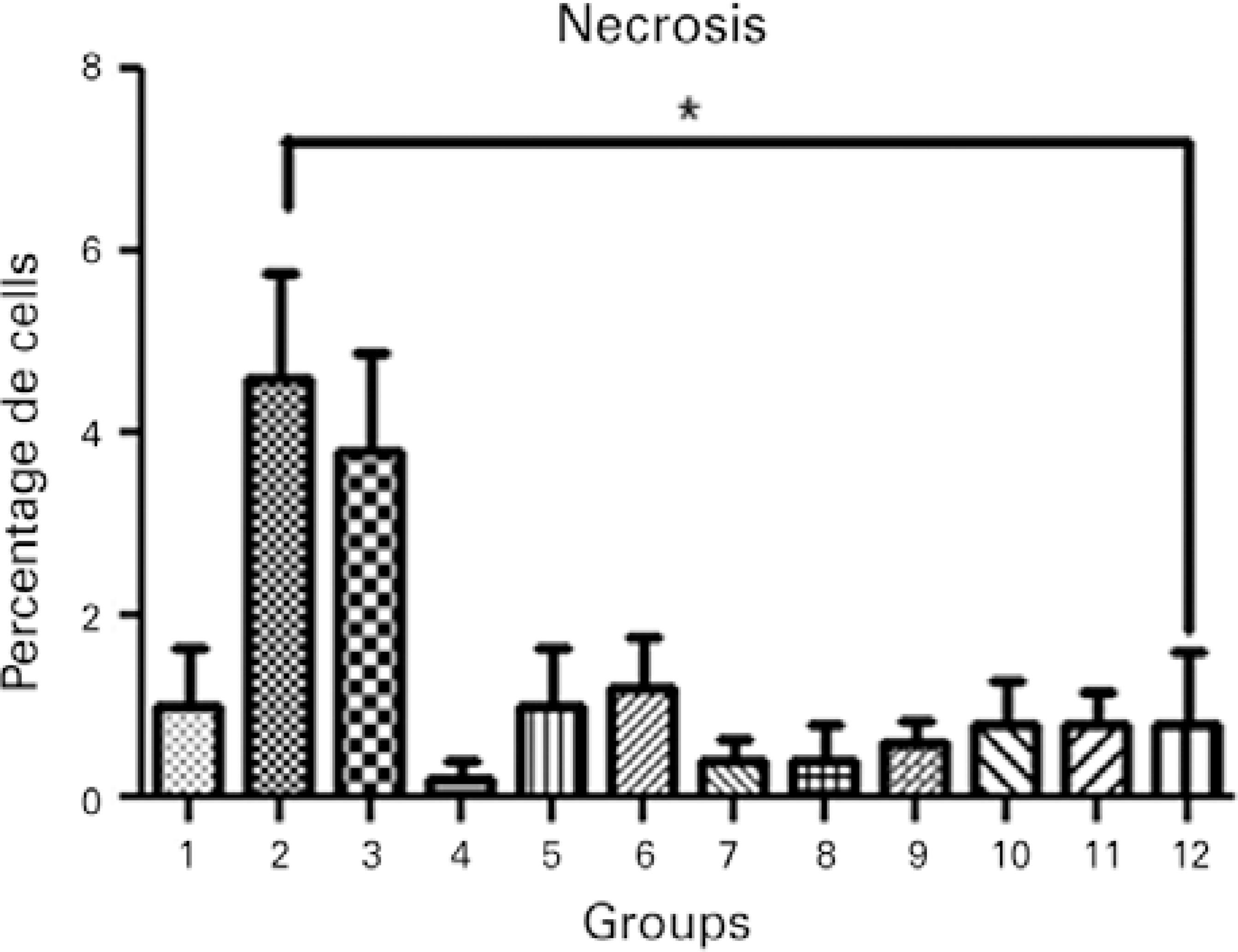
Figure 6 Necrosis in the different treatment groups: 1, keratocyte culture + riboflavin; 2, keratocyte culture + UVA light; 3, keratocyte culture + riboflavin + UVA light; 4, keratocyte culture + 200 µL of collagen + riboflavin; 5, keratocyte culture + 200 µL of collagen + UVA light; 6, keratocyte culture + 200 µL of collagen + riboflavin + UVA light; 7, keratocyte culture + 200 µL of collagen; 8, keratocyte culture + 500 µL of collagen + riboflavin; 9, keratocyte culture + 500 µL of collagen + UVA light; 10, keratocyte culture + 500 µL of collagen + riboflavin + UVA light; 11, keratocyte culture + 500 µL of collagen; 12, control group. All groups were treated for 30 minutes.Kruskal-Wallis test= P=0.007, Group 2 versus Group 12 (control); Dunn’s post-hoc test= P<0.05.
Immunofluorescence
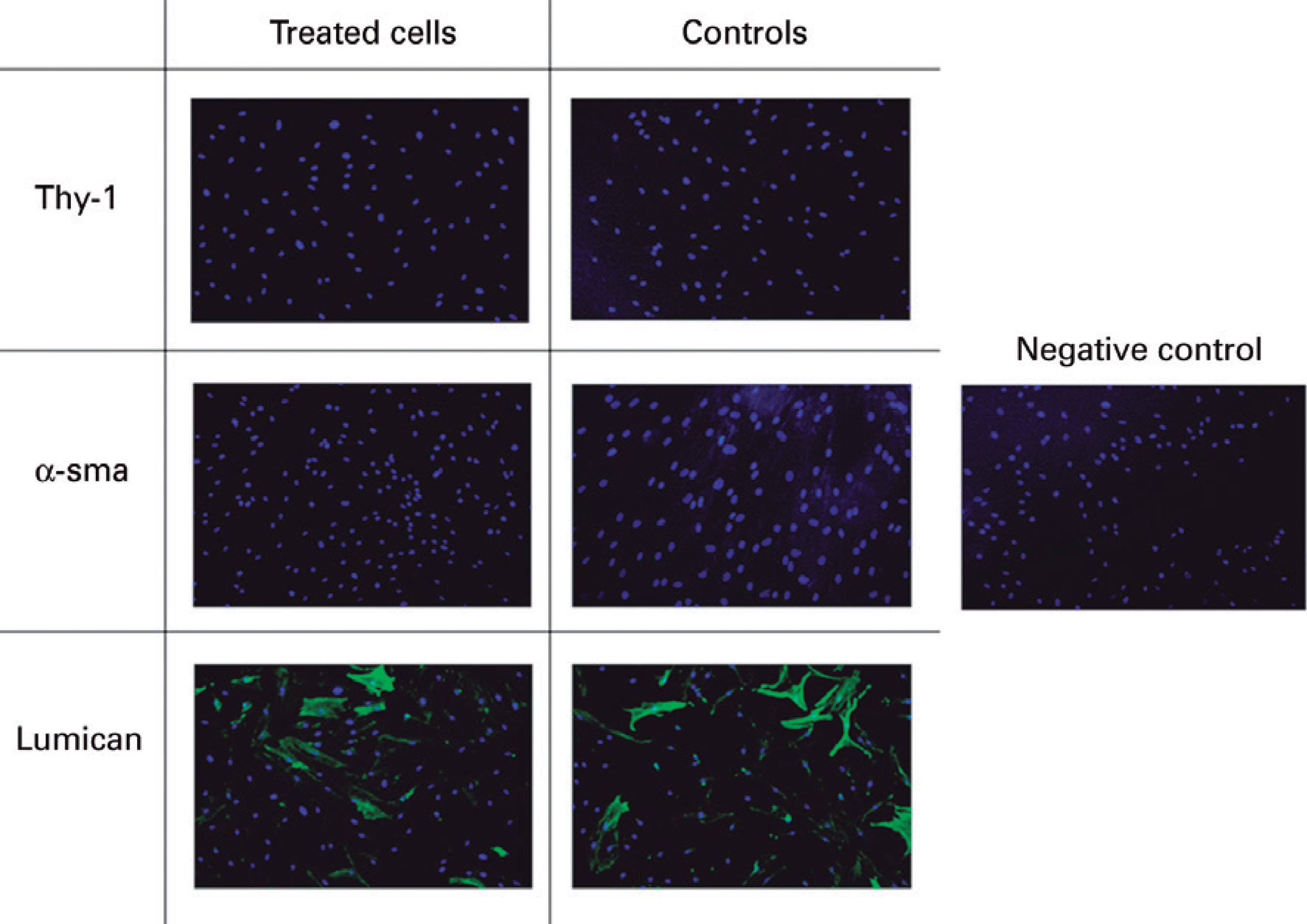
All cultures tested negative for the epithelial cell marker CK3, which was used to characterize cells (Figure 7). In all groups, cells were negative for fibroblast (Thy-1) and myofibroblast (α-sma) markers, and positive only for the keratocyte marker, (lumican), both before and following crosslinking (Figure 8). This staining pattern demonstrates the success of keratocyte culture, with staining remaining unaltered in all groups compared to the control group up to 24 hours after treatment.
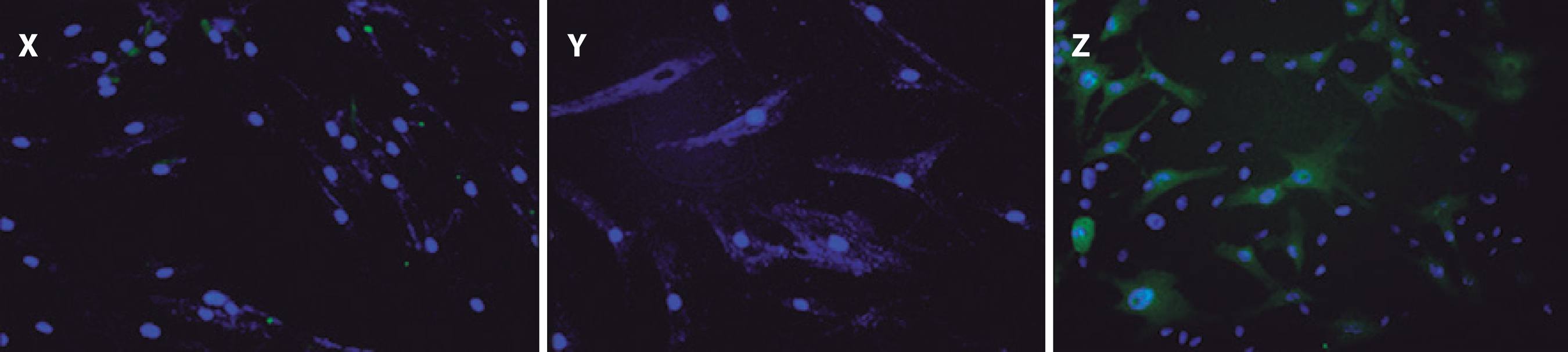
Figure 7 Immunofluorescence assay demonstrated negative expression of the differentiated corneal epithelial cell marker CK3 in cultured keratocytes. X) Immunofluorescence using CK3 antibody. Y) Negative control. Z) Positive control in cultured corneal epithelium.
DISCUSSION
The results from the present study indicate crosslinking does not cause severe damage to the human cornea because extensive cell death was observed only in keratocytes exposed to UVA light without the presence of collagen. In corroboration with previous reports, the present study also demonstrates that riboflavin has no cytotoxic effect. This finding was expected as riboflavin is a crucial component of all living cells and a precursor of flavin mononucleotide and flavin-adenine dinucleotide, two coenzymes essential for carbohydrate, fat, and protein metabolism present in the retina, liver, and heart(9-11).
To standardize keratocyte cultures, various experiments were required because the maintenance of cultured cells in the quiescent state was technically challenging because of high concentrations (10%-20%) of fetal bovine serum (FBS) promoting cell differentiation. FBS contains factors such as lysophosphatidic acid (LPA) and sphingosine-1-phosphate (S1P), which stimulate cell contractility and play a fundamental role in the transformation of quiescent keratocytes with dendritic morphology into cells with an activated fibroblast phenotype(12,13). On the other hand, the absence of FBS in culture medium hampers tissue adhesion to culture plates because culture medium is more fluid and facilitates cell detachment, thereby rendering cell growth impossible.
The findings of the present study may be clinically applicable as, to the best of our knowledge, no previous comparable studies using human keratocytes have been reported up to the present time.
The present study was performed using two-dimensional cultures to evaluate the effect of crosslinking by the addition of 200 or 500 µL of collagen to cell cultures, equivalent to the collagen thickness of the central and peripheral cornea, respectively. This research model partially reproduces the corneal stroma. 3 D culture models are generally considered to have greater model validity and to better mimic in vivo cell behavior(14,15) because keratocytes reside within a three-dimensional extracellular matrix. Therefore, 3 D models may allow more accurate observation of cell morphology, organization, and mechanical behavior(12). Furthermore, such culture systems may permit cell degradation and extracellular matrix remodeling to be investigated.
When culture medium with only 2% of FBS was used, optical analysis demonstrated that growing cells had a dendritic morphology similar to that of quiescent keratocytes. Cells were characterized by immunofluorescence analysis using the following markers: lumican, Thy-1, α-sma, and CK3. Lumican is one of the most abundant proteins in the corneal stroma and is expressed in the presence of quiescent keratocytes(16). Thy-1 has been shown to contribute to the transformation of corneal keratocytes into fibroblasts, with its expression posited to indicate the involvement of fibroblasts in the healing process(17). Alpha-smooth muscle actin (α-sma) is expressed during wound healing by fibroblasts undergoing transformation into myofibroblasts(18) with contractile activity that helps decrease the injured area(19,20). Finally, CK3 is expressed by differentiated corneal epithelium. Immunofluorescence of cultured cells in the present study demonstrated positivity for the lumican protein only, confirming that cultured cells were keratocytes in a quiescent state.
Immunofluorescence assays were performed after the cells were submitted to different treatments to assess for morphological modifications after exposure to different variables. Once again, cells cultured in these conditions expressed lumican only. In this step, the CK3 antibody, a differentiated corneal epithelium marker, was not used because its negative expression during cell characterization had already been demonstrated.
To confirm the results of the MTT colorimetric assays, the viability of keratocytes was evaluated using Hoechst 33342 and propidium iodide staining, allowing living cells and those in apoptosis and necrosis to be identified and quantified. Apoptosis is a highly regulated physiological process of programmed cell death and plays an important role in the homeostasis of different tissues in response to numerous stimuli. This process is characterized by various morphological and biochemical alterations that are of crucial importance for embryo development, for maturation of the immune system, and as a defense mechanism against external insults(21). During apoptosis, alterations occur in the cytoskeleton that induce the contraction and loss of cell volume, chromatin condensation, nuclear fragmentation, and the formation of cytoplasmic vesicles that give rise to structures termed apoptotic bodies(22). Necrosis is defined as a violent form of cell death initiated by environmental stimuli that results in rapid deregulation of homeostasis.
To validate the results of the present study, further studies using three-dimensional keratocyte culture with variations in the duration and intensity of UVA in vitro and in experimental animal models are required.
The present study evaluated the effects of riboflavin and UVA light, individually and in combination, on human corneal keratocytes in vitro. Cultures of keratocytes were successfully obtained and remained quiescent with dendritic morphology, as demonstrated by lumican immunofluorescence analysis. Immunofluorescence analysis of cells after exposure to riboflavin and UVA light demonstrated similar marker expression to untreated cells. MTT assays demonstrated lower cytotoxicity in groups containing collagen when exposed to UVA light. Staining with Hoechst 33342 and propidium iodide confirmed the results of MTT assays, with higher rates of apoptosis and necrosis observed in groups in which collagen was not added.

