INTRODUCTION
The condition of lacrimal glands (LG) is influenced by components of the immune, neural, and endocrine systems(1-5). Insulin is thought to be of major importance, based on observations related to its deprivation in diabetes mellitus (DM), on LG, tear film, and ocular surface in humans, animal models, and in culture(6-8). Lacrimal gland acinar cell (LGAC) culture is an appropriate model for improving the understanding of the influence of insulin on these cells.
In previous studies, the culture of LGACs from different species was performed over an average period of 21 days(9,10). Several attempts to enhance the growth of acinar cells in culture have been made, but after a span of approximately 3 weeks, LGACs exhibited decreased viability, with high numbers of apoptotic cells(11-14).
Exocrine acinar cells are fragile, post-mitotic epithelial cells with marked polarity. This indicates that the apical and basal sides are clearly positioned but also that intracellular organelles are located at one or the other pole, depending on their cellular functions. Thus, an adequate extracellular matrix and other specific ingredients are needed in the culture media to mimic in vivo conditions(15,16). The handling during isolation and culture procedure of LGACs should be gentle and kept to a minimum to avoid disturbing this organization and decreasing cell viability. Changes in culture medium, temperature, and time of handling decreases the amount and viability of isolated LGACs, resulting in unsatisfactory isolation and cell culture viability(9).
Rat LG acinar cells have to be handled carefully from isolation to culture. Also, a series of steps and parameters are necessary to separate acinar cells from the other LG cells (i.e., ductal, myoepithelial, endothelial, and neuronal cells and leukocytes)(17). To achieve this, it is necessary to apply binders and digestive enzymes. The selected cell populations are submitted to gradient centrifugation and filtering to isolate the target cells. The difficulty of this procedure not only pertains to cell isolation but also to obtaining good viability (higher than 80% of the total number of cells) to ensure a large population of target cells with preserved secretory activity(17).
Previous studies have focused on the aspects of LGACs in primary culture, applying standard amounts of insulin in the culture media(12,18). The aims of this study were to determine the optimal conditions for establishing a line of primary LGACs in culture and to study the effect of insulin in variable concentrations in the culture medium on acinar cell growth, secretory function, and viability.
METHODS
Animals and study design
Male Wistar rats (Rattus norvegicus , 8 weeks old) were euthanized to obtain LGACs. The study protocols and the project followed the guidelines of the Association of Research in Vision and Ophthalmology (ARVO) and were approved by the Ethics Committee on Animal Experimentation (CETEA) of the Faculty of Medicine of Ribeirão Preto, Universidade de São Paulo (FMRP-USP) (Process # 076/2004).
LGs were removed under intraperitoneal anesthesia with xylazine 15 mg/kg and ketamine 150 mg/kg. After harvesting LGs, the animals were euthanized by an excess of anesthesia. For each experiment, three animals were sacrificed, using both LGs of each animal, in a total of 15 experiments. Experiments with a cell viability of ≥80% after isolation and transfer of LGACs to culture plates were considered reproducible for data collection. A summary of the study flow is presented in figure 1.
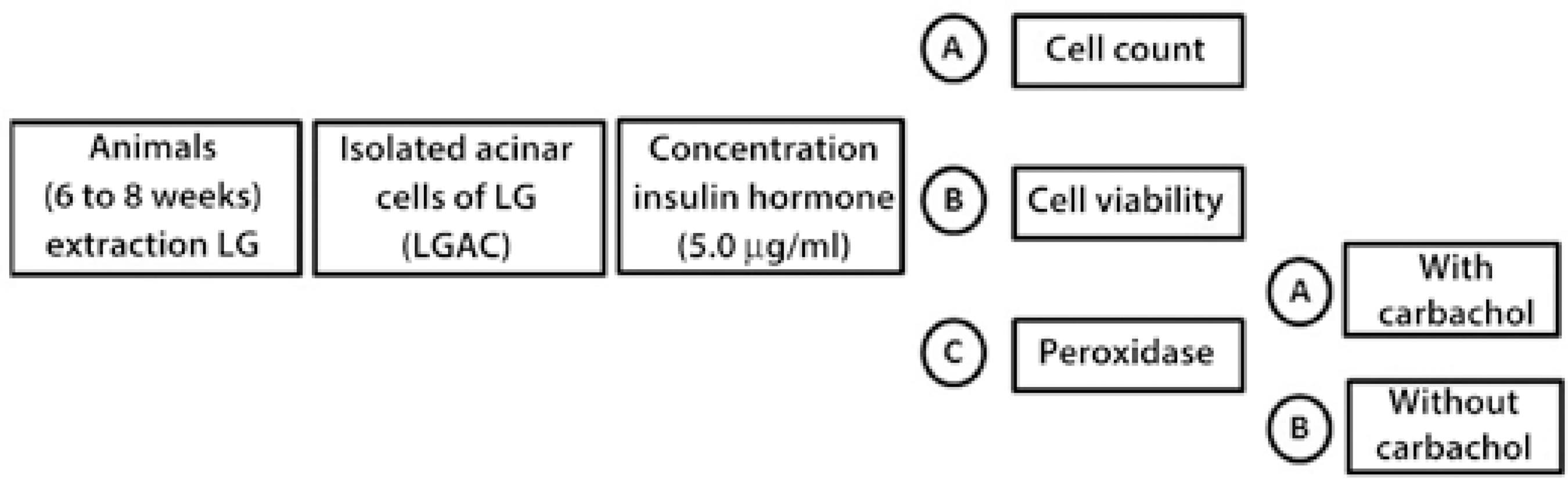
Figure 1 Schematic study design showing the steps involved in and timing of the evaluation of LGAC growth in primary culture.
All glassware and surgical instruments were cleaned with Extran MA 02 neutral (Merck KGaA, Darmstadt, Germany) and washed in Milli-Q® water (Thermo Fisher Scientific Inc, Barnstead Nanopure, Waltham, MA, USA) prior to sterilization and use for tissue harvesting, reagent preparation, and cell isolation.
Medium, reagents, and substrates
The culture medium was prepared based on the culture media used for LGAC cultures, as previously described(9,15,17), and was applied for LGAC isolation, cell manipulation, and cell culture.
For the preparation of the medium, 250 mL of low glucose (1 g/L) Dulbecco's Modified Eagles Medium (DMEM) (Invitrogen, Camirillo, CA) was supplemented with gentamicin 25 µg/mL, putrescine 1.0 mM, reduced glutathione 10 µg/mL, ascorbic acid 50 µg/mL, dexamethasone 10 µg/mL, insulin 5.0 µg/mL, selenium 5.0 ng/mL, transferrin 5.0 µg/mL, and carbachol 100 μM (Sigma-Aldrich, St. Louis, MO, USA). The reagents were carefully weighed (Bioprecisa, FA 2104N, PR, Brazil) and mixed under aseptic conditions in a fume hood (Nuaire, NU-425, USA) (Table 1).
Table 1 Composition of the supplemented low-glucose DMEM used in the primary culture of LG acinar cells
| Suplement | Final concentration |
|---|---|
| Putrescin | 1.0 mM |
| Reduced glutathione | 10.0 μg/ML |
| Ascorbic acid | 50.0 μg/ML |
| Dexamethasone | 10.0 μg/ML |
| Insulin | 5.0 μg/ML |
| Selenium | 5.0 μg/ML |
| Transferrin | 5.0 μg/ML |
After buffering the pH to 7.6 (Model 215, Denver Instrument Company, USA), the culture medium was filtered with disposable 22-µm sterile filters (Corning Inc, NY, USA) connected to a vacuum pump. Epithelial growth factor (EGF) was added (Sigma-Aldrich, St. Louis, MO) at a concentration of 10 ng/mL. The medium was stored at 4°C and removed 30 min prior to use to warm it up slowly to room temperature.
As specifically mentioned in the assays described below, the medium was supplemented with fetal bovine serum (FBS) (Invitrogen, Camirillo, CA, USA) or variable concentrations of insulin and carbachol (Sigma-Aldrich, St. Louis, MO, USA).
Cell isolation
After removal, each LG had its capsule gently removed and were washed in sterile petri plates (Techno Plastic Products TPP, Switzerland) with 1.0 mL of defined culture medium with 10 µL soybean trypsin inhibitor (Sigma-Aldrich, St. Louis, MO, USA). The glands were transferred to a new sterile petri plate (35 × 10 mm) (Corning, NY, USA) containing Hanks› balanced solution without Ca2+ and Mg2+ (HBSS, Invitrogen Camirillo, CA, USA), cut into small pieces, transferred to a 15-mL Falcon-type tube (Techno Plastic Products TPP, Switzerland), and shaken in HBSS with 0.5 M EDTA at 5 rpm for 15 min at room temperature (Arsec OSC1, Cotia, SP, Brazil). The glandular tissue was sedimented for 15 min and gassed with 95% O2 and 5% CO2 for 15 s.
After discarding the supernatant, the tissue was transferred to a spinner flask (Bellco Glass, Inc., NJ) with 5.0 mL culture medium with 10 µL hyaluronidase (150 U/mL), 25 µL collagenase (300 U/mL), and 25 µL DNAse (4 U/mL) (Sigma-Aldrich, St. Louis, MO, USA). Enzymatic digestion was performed using a magnetic stirrer (model 78 HW-I, Biomixer, Jiangsu, China) at 60 rpm at 37°C for 60 min. LGACs were separated by subsequent filtration with 100-µm and 70-µm cell strainers (BD Bioscience, NJ, USA). The decanted cells formed a pellet and were ready to culture.
Flow cytometry
After centrifugation at 5000 rpm for 15 min at room temperature (Eppendorf 5417 R, Hamburg, Germany), the pellet had clearly segregated into two phases: a thick, light brown phase on the bottom and a light red phase on the top (Figure 2). Samples of 10 µL of LGAC suspension (n=3) were gently obtained by aspiration with a micropipette, (Eppendorf, Hamburg, Germany) without mixing the phases, and diluted in a phosphate buffer. These samples were subjected to flow cytometry, using fluorescence-activated cell sorting (FACS) in triplicate samples (BD FACS Array FACS Diva version 6.1.1 software, BD Bioscience, NJ, USA) to confirm the predominance of LGAC upon isolation. Rabbit IgG antibody against the cell membrane water channel aquaporin-5 (Anti-AQP5, Sc-28628, Santa Cruz Biotechnologies, Santa Cruz, CA, USA) was applied because its expression in LGs is limited to acinar cells(19).

Figure 2 Illustrative photos of the key steps involved in LGAC primary culture. A) Rat exorbital lacrimal gland isolated (arrow). B) Lacrimal gland cutting in a Petri dish and embedding of small fragments in culture medium. C) Enzymatic digestion of the lacrimal gland on a magnetic stirrer using a spinner flask. D) Lacrimal gland cell pellet after centrifugation (arrow). E) LGAC counting and viability testing using a Neubauer chamber and trypan blue after isolation.
The following conditions were compared: cells obtained from the bottom and cells obtained from the top of the pellet, following cell separation and centrifugation. The "bottom cells" were assumed to be acinar cells (heavier), and the "top cells" were assumed to be fibroblasts, myoepithelial, and endothelial cells (lighter)(15). The percentage of acinar cells (positive for aquaporin-5) was compared in samples from the bottom and top of the pellet.
Samples of LGAC suspension were blocked with autologous rat serum and incubated for 1 h with the primary antibody (Anti-AQP5) at 4 µg/mL, followed by 30 min of exposure to the secondary antibody (goat anti-rabbit IgG-FITC, Sc-2012 Santa Cruz Biotechnologies, Santa Cruz, CA, USA) at 2.5 µg/0.5 mL, as previously described(18).
Cell culture
The acinar cells were cultured in 24-well culture plates (Techno Plastic Products TPP, Switzerland) pre-coated with MatrigelTM (BD Bioscience, NJ, USA). To each well, 200 µL of cell suspension (an average of 3 × 105 cells) and 300 µL of culture medium were added.
Other culture approaches were attempted, including leaving the plastic bottom without any coating or coating with poly-L-lysine or fibronectin (Sigma-Aldrich, St. Louis, MO, USA) instead of Matrigel.
Cultures were terminated at specific time points. The medium was replaced by 300 µL of culture medium containing trypsin (1.0 mg/mL) and EDTA (0.4 mg/mL) (Sigma-Aldrich, St. Louis, MO, USA) and incubated at 37°C and 95% O2 and 5% CO2 for 45 min (Nuaire, N4750 G, USA).
Prior to and after culture termination, cell counting and viability were performed by analyzing 10 μL of cell suspension in culture medium mixed with 10 µl of 2% trypan blue (Sigma-Aldrich, St. Louis, MO, USA) using a Neubauer chamber (Optik Labor, Germany).
As mentioned above, data on cell numbers and cell viability of primary culture LGACs cultivated under different conditions were obtained. Culture media and cells were stored separately at -80°C (So-Low-Ultra-Low Freezer, Cincinnati, OH, USA) for subsequent biochemical analysis.
Peroxidase activity assay
A peroxidase activity assay was performed with aliquots of 50 µL of LGAC primary culture samples using a commercial kit for peroxidase (Invitrogen Camirillo, CA, USA) according to the manufacturer›s protocol (n=5 wells for each condition; samples tested in duplicates). Light absorbance was observed using a spectrophotometer (SpectraMax M5, San Diego, California, USA).
To assess the influence of insulin, levels of insulin in the culture medium during the culture process were maintained as usual (5.0 μg/mL), 10 times higher (50 μg/mL), or 10 times lower (0.5 μg/mL).
Further, 100 mM carbachol was added to the culture medium 20 min prior to culture termination to compare peroxidase secretory capacity over time and in response to different insulin concentrations in the culture media.
RESULTS
Analysis of LGAC populations isolated and separated by FACS from the two pellet layers (top and bottom) showed that the cells isolated from the bottom layer represented 20% acinar cells on average; meanwhile, samples from aliquots obtained from the top of the pellets had less than 3% cells positive for aquaporin-5 (acinar cells), confirming the efficiency of the method of cell isolation (Figure 3).
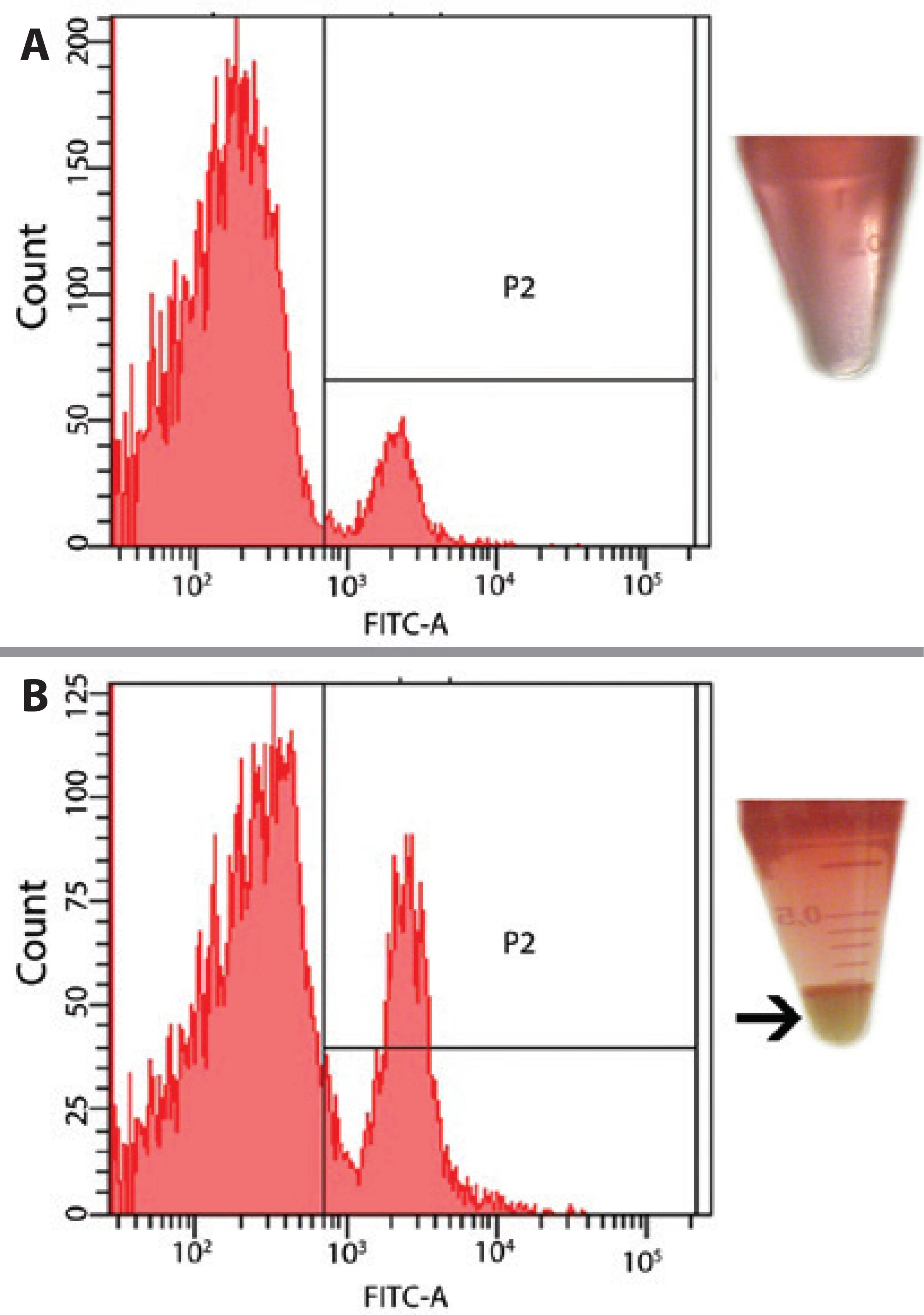
Figure 3 A) A low amount of aquaporin-5-positive LGACs (3%) was observed in the supernatant. B) A higher amount of positive cells (20%) was observed in the pellet (arrow), indicating the efficiency of the isolation method.
By the fourth day, Matrigel proved to be a superior substrate for LGAC growth with respect to cell shape and number of viable cells compared with plastic, poly-L-lysine, and fibronectin (Figure 4). The cells growing on Matrigel were larger, and the number of cells was similar compared with that at day 0.
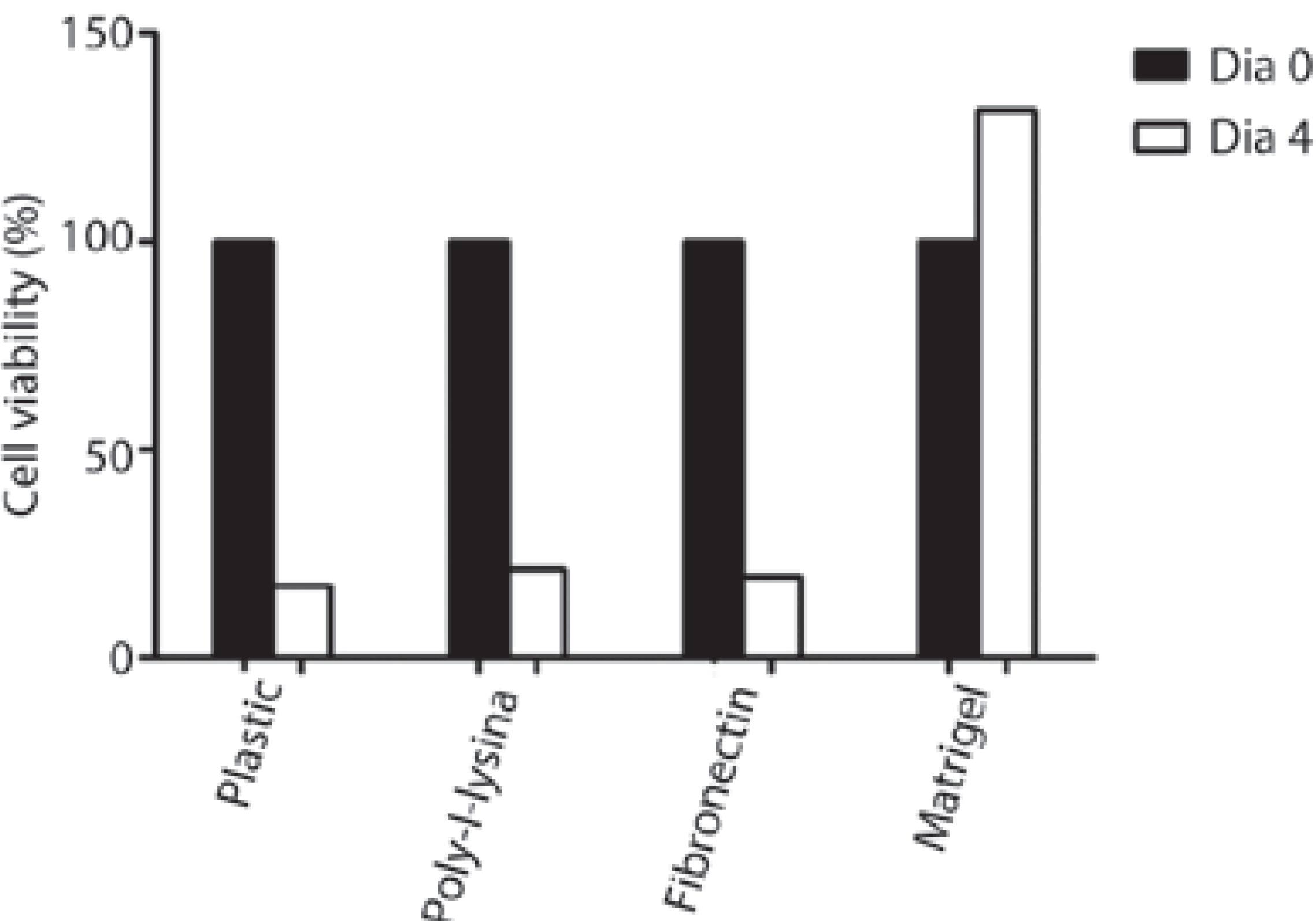
Figure 4 Counts of LG acinar cells in primary culture on four consecutive days, using supplemented low-glucose DMEM. Comparative analysis of four substrates: plastic, poly-L-lysine, fibronectin, and Matrigel at day 0 (black columns) and day 4 (white columns) (n=3 wells/condition).
The time length of the survival curve of LGACs in primary culture with supplemented low-glucose DMEM on Matrigel-covered culture plates was evaluated. The mean levels, as measured every 3 days, revealed a higher number and proportion of viable cells at day 3, with a continuous decline until day 12 (Figure 5).
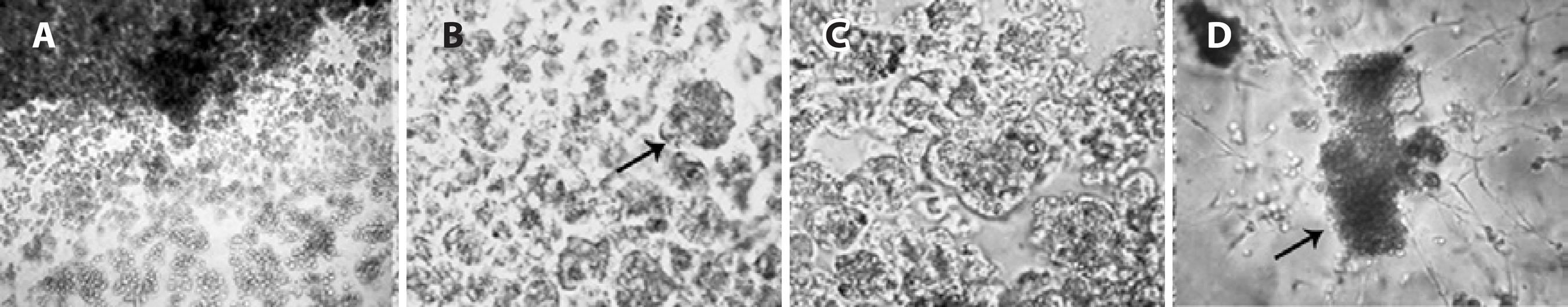
Figure 5 Microscopic aspects of LG acinar cells in culture on day 4. A) The wave front of cell proliferation by the third day (100x magnification). B) LG cells forming clusters similar to acinar units (arrow) by the third day (200x magnification). C) Structures compatible with secretory granules in the cytoplasm (arrow) were observed on the fifth day (200x magnification). D) Different morphologic types of LG cells, suggesting the proliferation of remaining myoepithelial and mesenchymal cells by the third week (400x magnification).
Daily observation of LGACs in culture by microscopy revealed, from the third to the fourth day, the wave front of cell proliferation; it was also possible to observe cells attaching to each other, forming clusters similar to acinar units, and in certain cells, structures compatible with secretory granules were observed in the cytoplasm. Longer periods of LG culture revealed different morphologic types of cells in parallel with a reduction in the number of acinar cells between days 21 and 28 of culture, suggesting the proliferation of residual myoepithelial and mesenchymal cells (Figure 6).
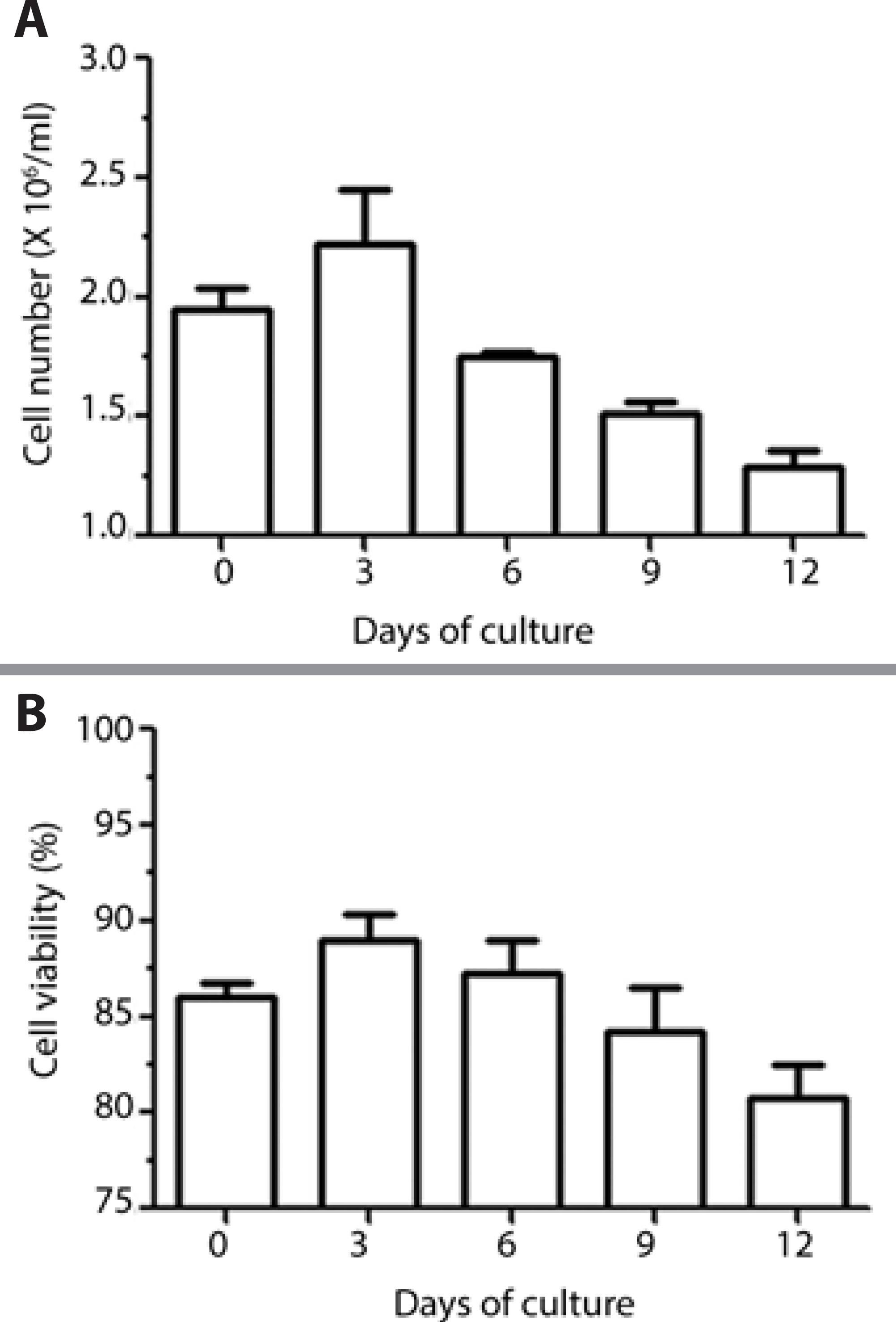
Figure 6 Counts of LG acinar cells in primary culture over a span of 12 days, using supplemented low-glucose DMEM and Matrigel substrate; comparative analysis of cell number (A) and viability (B) every 3 days, from the seeding day (0) until day 12 (n=3 wells/condition).
To investigate the secretory capacity of the isolated acinar cells in culture, the peroxidase activity was measured in the supernatant prior to and after stimulation with 100 µL of 100 mM carbachol. The data showed that the peroxidase secretion, as measured in the medium at days 3 and 6, was efficiently stimulated by carbachol. However, these returned to non-stimulated levels (indifferent to cholinergic stimulation) by day 9 (Figure 7A).
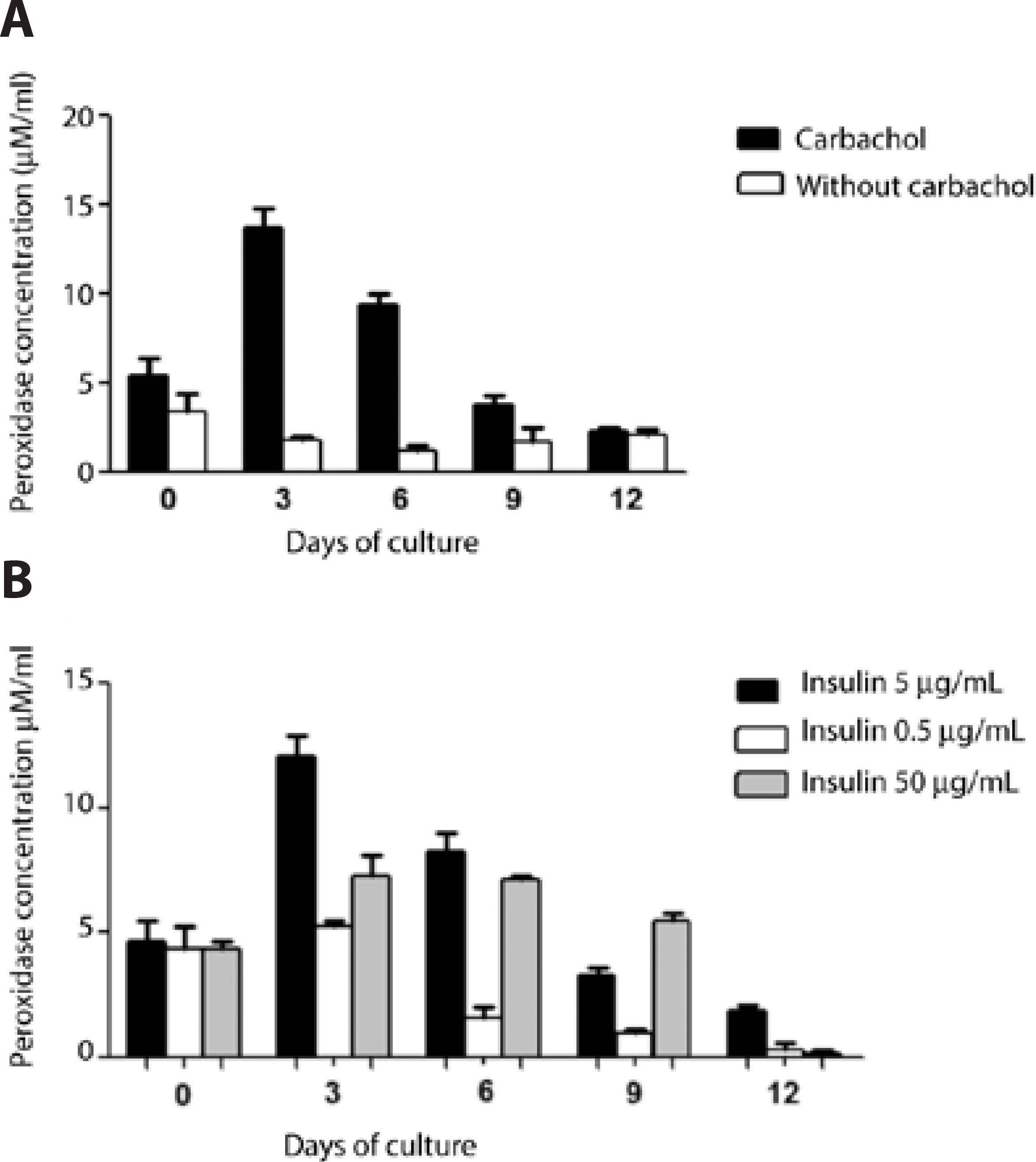
Figure 7 Peroxidase activity in the culture medium using Matrigel substrate of LG acinar cells in primary culture from day 0 to 12. A) The culture medium containing the standard insulin concentration (5.0 μg/mL) was collected at the indicated days, prior to and 30 min after incubation with 100 mM carbachol. Peroxidase activity was analyzed as described in the methods (n = 2-4 samples/condition). B) The effect of insulin concentration on secretory activity over a period of 12 days in response to 100 mM carbachol stimulation. LGACs were cultured using three concentrations of insulin (0.5, 5.0, and 50.0 μg/mL). The culture media was collected on the indicated days (days 0-12) and peroxidase activity was measured (n=3/condition in duplicates) as described in the methods section (black filled bars, 5.0 μg/mL insulin; white filled bars, 0.5 μg/mL insulin; grey filled bars, 50.0 μg/mL of insulin).
To determine changes in cell proliferation due to changes in the concentration of insulin in the supplemented low-glucose DMEM, analysis of cell count and viability in experiments using three different insulin concentrations (0.5, 5.0, and 50.0 μg/mL) was performed. The data showed that at day 0 and day 3, there was no change in cell number or cell viability; however, at day 6, the acinar cells exposed to lower insulin concentrations began to decline in viability, and after day 9, both the number of cells and viability of the culture using supplemented low-glucose DMEM with 0.5 and 50.0 μg/mL of insulin presented relatively lower numbers (Figure 8).
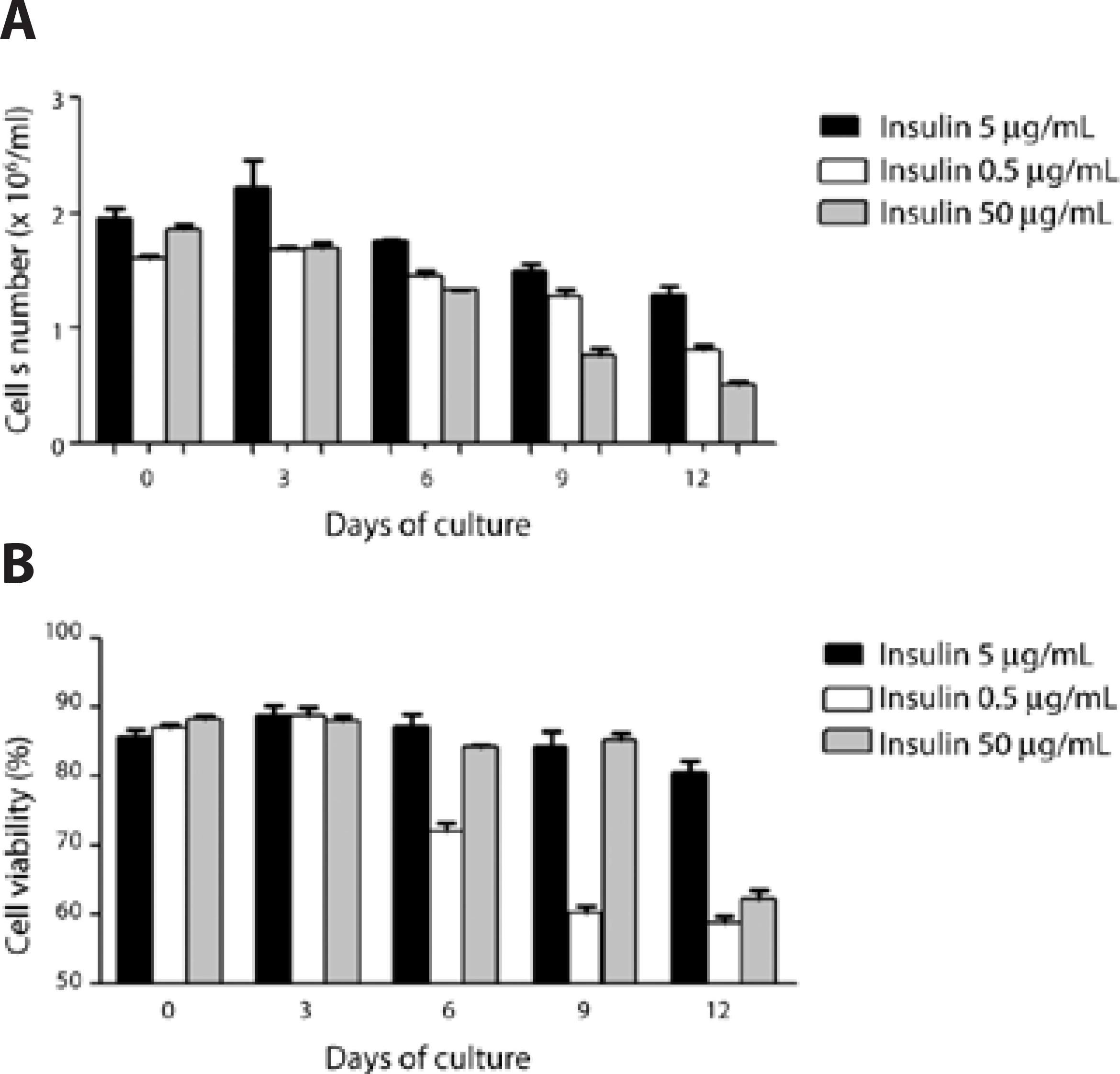
Figure 8 The effect of insulin concentration in the supplemented low-glucose DMEM media on LGAC proliferation in primary culture, using Matrigel substrate over a span of 12 days. Comparative analysis of cell number (A) and viability (B) in response to three concentrations of insulin in the medium (0.5, 5.0, and 50.0 μg/mL) was evaluated. The culture was discontinued at the indicated days (0 through 12), and cell counting and viability testing was performed (n=3/condition) using a Neubauer chamber and trypan blue staining (black filled bars, 5.0 μg/mL insulin; white filled bars, 0.5 μg/mL insulin; grey filled bars, 50.0 μg/mL of insulin).
Comparative analysis of the secretory activity under different insulin conditions was performed by stimulation with 100 µL of 100 mM carbachol for 30 min and evaluating the peroxidase activity in samples obtained from the supernatant from cultures with three different concentrations of insulin (0.5, 5.0, and 50 μg/mL) every 3 days (from day 0 to day 12). The data showed that insulin at a concentration of 5.0 μg/mL supported a higher peroxidase secretion from LGAC, measured by its activity in the culture medium, than insulin at a concentration of 0.5 μg/mL; this finding was most remarkable at day 3. The decline in peroxidase secretion over the 12 days was more prominent in the hypoinsulinic medium (0.5 μg/mL) and similar between the standard and hyperinsulinic media (5.0 and 50.0 μg/mL, respectively; Figure 7 B).
DISCUSSION
The present study introduced a feasible protocol for the isolation and culture of LGACs. Moreover, it elucidated aspects pertaining to the longevity of primary culture and the influence of insulin on culture maintenance.
This information contributes to the understanding of the role of LG homeostasis, its relationship with the endocrine system, and the potential effects of LG on the ocular surface.
The cell isolation procedure in this report included a reduction of several of the usual steps, such as repeated washings and centrifugations, and the use of Ficoll to isolate LGAC, as previously described(17). The simplified procedure did not compromise the purity of LGAC cells and helped to increase the longevity of the primary culture, compatible with recently published protocols(10,11). Our hypothesis is that reduced manipulation during isolation contributes markedly to improving and extending cell viability and behavior during culture.
Moreover, removal of the fibrous capsular tissue that surrounds LGs proved to be a crucial step for optimizing isolation of LGACs and thus successful culture.
An increase in both the number of living cells and cell viability with insulin at 5.0 μg/mL was observed from the third to the ninth day of cultivation, compared with day 0 (seeding), as well as with lower or higher doses of insulin, indicating a key role of insulin in a certain range for the homeostasis and proliferation of LGAC(8,20).
The trend to form clusters after the third day revealed that these cells show polarity, as previously observed(15). In some of the experiments, the cell culture was extended to investigate the behavior of LGACs in culture plates.
The method for isolating LGAC is efficient with respect to excluding most other cell types, as observed in the flow cytometry assay with aquaporin-5, where the number of AQP5-positive LGACs was substantially higher. The fact that more than 60% of the cells had not been detected by flow cytometry may be explained by a weak antibody affinity. Immunocytochemistry analyses at different stages of serial cultures could give an indication of cell lines involved during culture(18).
Fibroblasts appeared by the third week of culture. None of these observations had been registered in our previous experiments or reported by other authors in studies using shorter culture periods(10,17,18,21).
The use of a combination of quinolone (ofloxacin) and aminoglycoside (gentamicin) was effective in terms of preventing contamination of the culture. One of the experiments failed because of the presence of filamentous fungi. It is assumed that an anti-fungal drug is necessary to prevent this issue.
None of the assays achieved cell culture confluence; therefore, neither passages nor sub-cultures were performed.
A relevant item that has not been highlighted previously with respect to maintaining LGAC cultures is the addition of carbachol in doses one thousand times less (100 μM) than those used to stimulate secretion. It is probably necessary to support these cells through neurotrophic pathways, as recently observed in embryonic salivary acinar cells, to enhance growth and differentiation(22).
Higher doses of carbachol (100 mM) were useful to confirm the secretory capacity of LGACs in culture. The secretory product evaluated was the enzyme peroxidase, which is present in tears and is linked to defense functions(23,24). Our observations revealed a time-dependent secretory capacity of these cells in culture with a decline after day 6, which also depended on insulin levels. Higher levels of insulin in the medium did not improve or extend the period of peroxidase secretion, which may be explained by faster consumption of glucose and/or achieving the limit of cell-specific metabolic activity.
In conclusion, this study introduced a method for simplifying isolation of LGACs using a cell strainer and a cell spin flask. In the culture method, we highlighted the importance of Matrigel as a culture substrate and low concentration of carbachol in the medium to maintain cell tropism. Also, most importantly, we identified the requirement for and the key concentration of insulin in the culture medium to maintain LGAC growth and secretory function. Improved understanding of culture methods can help develop strategies to restore post-mitotic injured tissues in the context of regenerative medicine, specifically in tissue engineering.

