INTRODUCTION
Arthur Alport first published his description of a triad of symptoms in a family with hereditary congenital hemorrhagic nephritis, deafness, and ocular changes in 1927. To this day, Alport syndrome may still inevitably leads to end-stage renal disease and the need for renal replacement therapy, starting in young adulthood(1). Alport syndrome has a prevalence of 1 in 5,000, and 85% of patients have the X-linked form, which mainly affects males. The typical ocular associations are a dot-and-fleck retinopathy that occurs in approximately 85% of affected adult males, anterior lenticonus that occurs in approximately 25%, and the rare posterior polymorphous corneal dystrophy (PPCD). The retinopathy and anterior lenticonus do not usually appear during childhood but tend to worsen with time so that the retinal lesions are often present at the onset of renal failure; the anterior lenticonus develops later. The presence of anterior lenticonus or posterior polymorphous corneal dystrophy in any individual is highly suggestive for the diagnosis of Alport syndrome. Additional ocular features described in X-linked Alport syndrome include other corneal dystrophies, microcornea, arcus juvenilis, iris atrophy, cataracts, spontaneous lens rupture, spherophakia, posterior lenticonus, a poor macular reflex, fluorescein angiogram hyperfluorescence, electrooculogram and electroretinogram abnormalities, and retinal pigmentation. All mutations that have been detected to date in X-linked Alport syndrome have affected the COL4A5 gene, which encodes the alpha-5 chain of type IV collagen. This protein plays a role in the basement membranes of the glomerulus, cochlea, retina, lens capsule, and cornea, resulting in abnormal ultrastructures(2).
CASE REPORT
RCBL, a 33-year-old female, with previously diagnosed Alport syndrome, was referred to the contact lenses sector complaining of poor vision in OU. She had presented chronic renal failure (two kidney transplantations), and had undergone cataract surgery in both eyes one year ago due to anterior lenticonus (Figure 1-3).

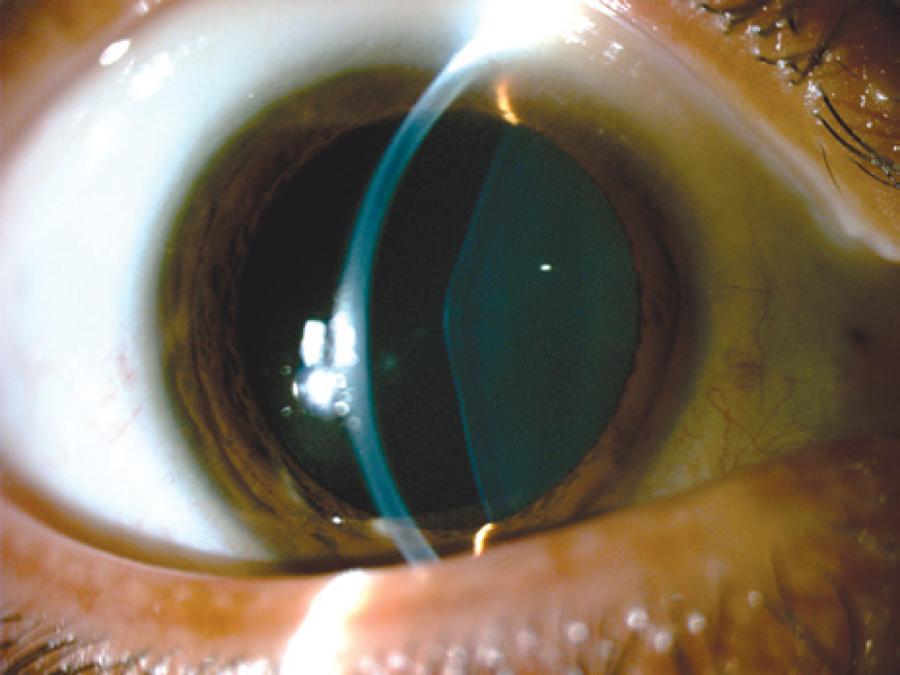
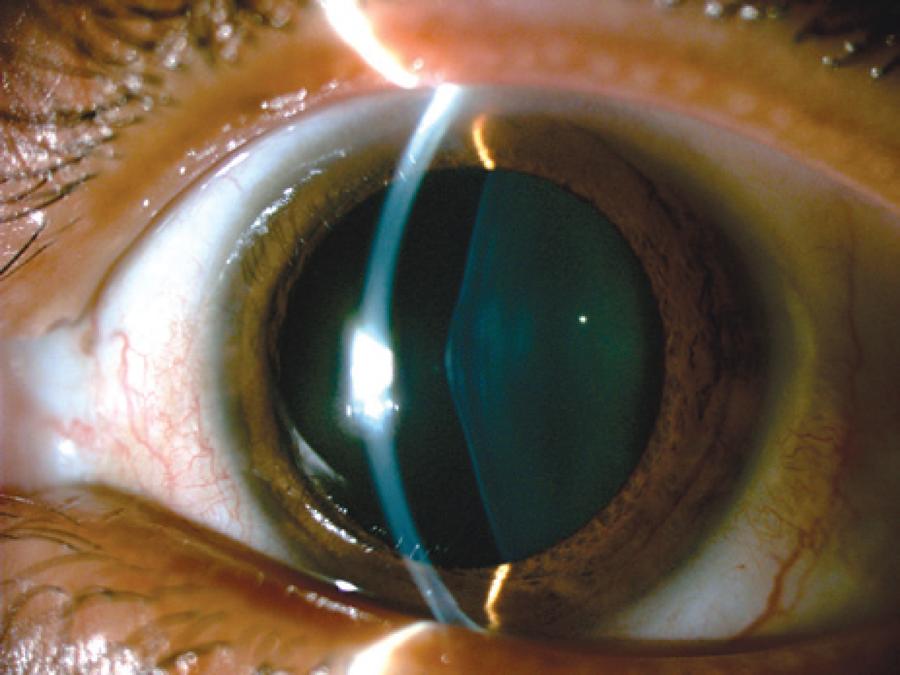
Ophthalmological examination denoted bilateral posterior polymorphous dystrophy (PPD), intraocular lenses that were in the bag in both eyes, as well as multiple perimacular changes on fundoscopy (dot-and-fleck retinopathy). Her refraction was OD: -7.00 -1.00 @ 180 (VA: 0.30 - LogMAR) and OS: -3.25 -4,00 @ 125 (VA: 0.40 - logMAR). Her corneal topography showed an irregular astigmatism with superior steepening in both eyes (Figure 4).
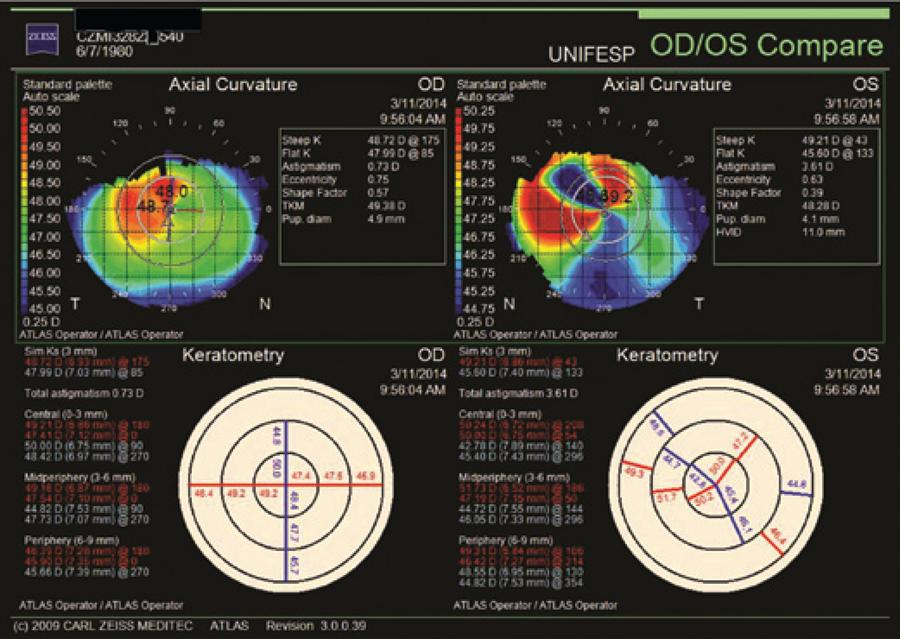
Figure 4 Corneal topography, after cataract surgery, shows irregular astigmatism with superior steepening OU.
After comprehensive ophthalmological examination and rigid gas permeable contact lens fitting, we were able to improve her visual acuity to 0.18 in OD and 0.30 in OS (logMAR). The prescribed contact lens had the following parameters:
DISCUSSION
The main features of Alport Syndrome are kidney failure, hearing loss, and ocular features, such as anterior lenticonus (15%-25% of patients, and may also be associated with cataract), dot-and-fleck retinopathy (85%), and PPCD(3). Aldaveet al suggested that there was a gene (ZEB1 ) involved in keratocyte function that may play a role in both the corneal stromal and endothelial development and function. The identification of abnormally steep corneal curvatures in 37% of all individuals with PPCD and in 86% of affected individuals with PPCD secondary to ZEB1 mutations confirms this association(4). Many genetically determined corneal dystrophies can occur in association with keratoconus including PPCD, lattice dystrophy, granular dystrophy, and Fuchs endothelial dystrophy. The association with so many corneal dystrophies and ocular abnormalities implies either a similarity in the underlying genetic defect or a tightly-linked network of interacting proteins, with a final common developmental pathway(5). PPCD and keratoconus share a potential common area of involvement (the posterior corneal surface, specifically Descemet's membrane) and/or an underlying commonality for the pathophysiology of corneal dystrophies. A recent paper characterized the cornea in patients with PPCD and demonstrated that the topographic parameters were significantly steeper but had no clinical or topographical evidence of keratoconus. The authors accumulated a cohort of patients who presented between 1982 and 2004 with a mean topographic simulated keratometry reading of 52.21 D, ranging from 46.47 D to 59.86 D, with no slit lamp or topographic findings suggestive of keratoconus(6). In this particular case, our patient had Alport Syndrome, PPCD, irregular astigmatism with superior steepening in both eyes; however, her visual acuity was improved with rigid gas permeable (RGP) scleral CLs despite other findings.

