INTRODUCTION
Since Stocker et al.(1) established a human corneal endothelial cell (HCEC) culture, the potential for cell therapy to treat corneal endothelial dysfunction using HCECs has demonstrated continuous development(2-8). The limitations associated with this therapy can be basically divided into two major areas: those related to culturing the cells, such as proliferation, cellular senescence, and fibroblastic transformation and those related to the logistics and techniques for transplanting the cells(2-8).
Regarding the first area, we recently published an article demonstrating the applicability of human serum-supplemented medium (HS-SM) as an alternative to fetal bovine serum-supplemented media (FBS-SM), suggesting potential advantages of a medium with fewer animal-derived components(9). Regarding the second area, the cryopreservation of both corneas for transplantation(10) and cultured HCEC using a standard medium (FBS-SM) have been already demonstrated(11) and could potentially improve the logistics related to the distribution of cultured HCECs for clinical use. The purpose of this study is to determine the ability of HCECs grown in HS-SM to be cryopreserved and maintain their characteristics compared with cryopreserved cells grown in FBS-SM.
METHODS
This study adhered to the tenets of the Declaration of Helsinki.
Materials were obtained from the following sources: serum-free medium (OptiMEM I), Dulbecco phosphate-buffered saline, Hank's balanced salt solution (HBSS), gentamicin, trypsin-EDTA, and goat serum (Gibco BRL/Life Technologies, Rockville, MD); mouse submaxillary gland nerve growth factor (NGF), human epidermal growth factor (hEGF), and bovine pituitary extract (BPE) (Biomedical Technologies, Stoughton, MA); FBS (Hyclone, Logan, UT); heat-inactivated human serum (HS, Valley Biomedical, Winchester, VA); ascorbic acid, chondroitin sulfate, calcium chloride, 0.02% ethylene diamine tetra acetic acid (EDTA) solution, and antibiotic/antimycotic solution (Sigma, St. Louis, MO); cell attachment reagent (FNC Coating Mix; Athena Environmental Sciences Inc., Baltimore, MD); dimethyl sulfoxide (DMSO) (Mediatech Inc, Manassas, VA); rabbit polyclonal antibody to ZO-1 (Invitrogen, Camarillo, CA); mouse monoclonal antibody to Na+/K+-ATPase alpha 1 subunit (Santa Cruz Biotechnology, Dallas, TX); mouse monoclonal antibody to glypican 4 (GPC4) and donkey serum (Novus Biologicals, Littletown, CO); AlexaFluor 488 donkey anti-mouse IgG (Life Technologies, Eugene, OR); AlexaFluor 555 goat anti-mouse IgG and Hoechst nuclear staining (Molecular Probes, Eugene, OR); and eight-well glass chamber slides (Laboratory-Tek, Nunc, Rochester, NY).
Human corneal endothelial cell culture
Three pairs of human corneas from donors aged 8, 28, and 31 years (referred to as donor 1, 2, and 3, respectively) were obtained from Lions VisionGift (Portland, OR) and the Alabama Eye Bank (Birmingham, AL). Donors had died from trauma, cardiomyopathy, and pneumonia, respectively, and death-to-preservation time was 4, 6, and 8 h, respectively. Standard eye bank protocols for informed consent and protection of donor confidentiality were used. Donor corneas were unsuitable for transplantation. Standard serologies for infectious agents were negative for the three donors. Primary cultures were initiated within 2-10 days of preservation in Optisol-GS.
Culture media
FBS-SM was previously described in a study(12) and was used in our laboratory(11) and comprised serum-free medium (OptiMEM-I), 8% FBS, 5 ng/mL hEGF, 20 ng/mL NGF, 100 mcg/mL BPE, 20 mcg/mL ascorbic acid, 200 mg/L calcium chloride, 0.08% chondroitin sulfate, 50 mcg/mL gentamicin, and antibiotic/antimycotic solution (diluted 1/100). HS-SM was described in our previous study(9) and comprised the same components as FBS-SM, with the exception that 8% FBS was replaced with 8% HS. The concentration of serum (both in FBS and HS) that was used to culture cells in glass chamber slides was doubled to 16%.
Isolation and growth of HCECs
HCEC populations were established according to published methods(12). Corneas were removed from corneal preservation media (Optisol-GS, Bausch & Lomb Inc., Rochester, New York), and Descemet membranes (DM) with intact endothelium were peeled. From each pair of corneas, one DM was used to initiate an HCEC culture with HS-SM and the other DM was grown with FBS-SM, comprising a total of six cell populations. DM was placed in either FBS- or HS-SM for 24 h at 37°C under 5% CO2. Next, the tissues were incubated in 0.02% EDTA for 1 h at 37°C to loosen cell-to-cell junctions and were manually disrupted by a passage through a pipette. The dissociated cells were centrifuged at 3000 rpm for 5 min, and the pellet was gently re-suspended in the respective medium. Isolated cells and pieces of DM were plated in a 6-well tissue culture plate that was pre-coated with undiluted cell attachment reagent (FNC Coating Mix). Initial plating density (P0) was variable and depended on the number of cells recovered from each DM. Overall, we would estimate the initial plating density as being between 100-200 cells/mm2 (approximately 104 to 2 × 104 cells/well of a 6-well plate). Cultures were then incubated at 37°C in a humidified 5% CO2 incubator, and the medium was changed every other day.
Cryopreservation of HCECs
At the day when the primary cultures [passage (P) 0] reached confluence, cells were trypsinized and re-suspended in their respective culture media (HS- or FBS-SM). They were then gently centrifuged and re-suspended in freezing medium comprising 90% initial medium and 10% DMSO. The cells were then slowly frozen overnight in an isopropanol bath freezing container at -80°C and subsequently transferred to liquid nitrogen. For recovery from cryopreservation, all cells were thawed at different time points (1, 2, and 3 months for donor 1, 2 and 3, respectively) in a water bath at 37°C, and the cells were promptly re-suspended drop-by-drop with their respective pre-warmed culture medium. The cells were then centrifuged, washed, and re-suspended in the culture medium to remove residual DMSO and were plated on pre-coated standard tissue culture plates (each cell population was seeded in two wells of a 6 well-plate). Subsequently, cells were passaged (P1-P5) on the day when they reached confluence, except for those used for immunofluorescence, which were maintained for 3 additional days after reaching confluence. To identify each cell population at each time point, the cells were named according to the passage by which they had been generated (e.g., P1 cells originated from the first passage). To enhance the understanding of the methodology, the process is shown in figure 1.
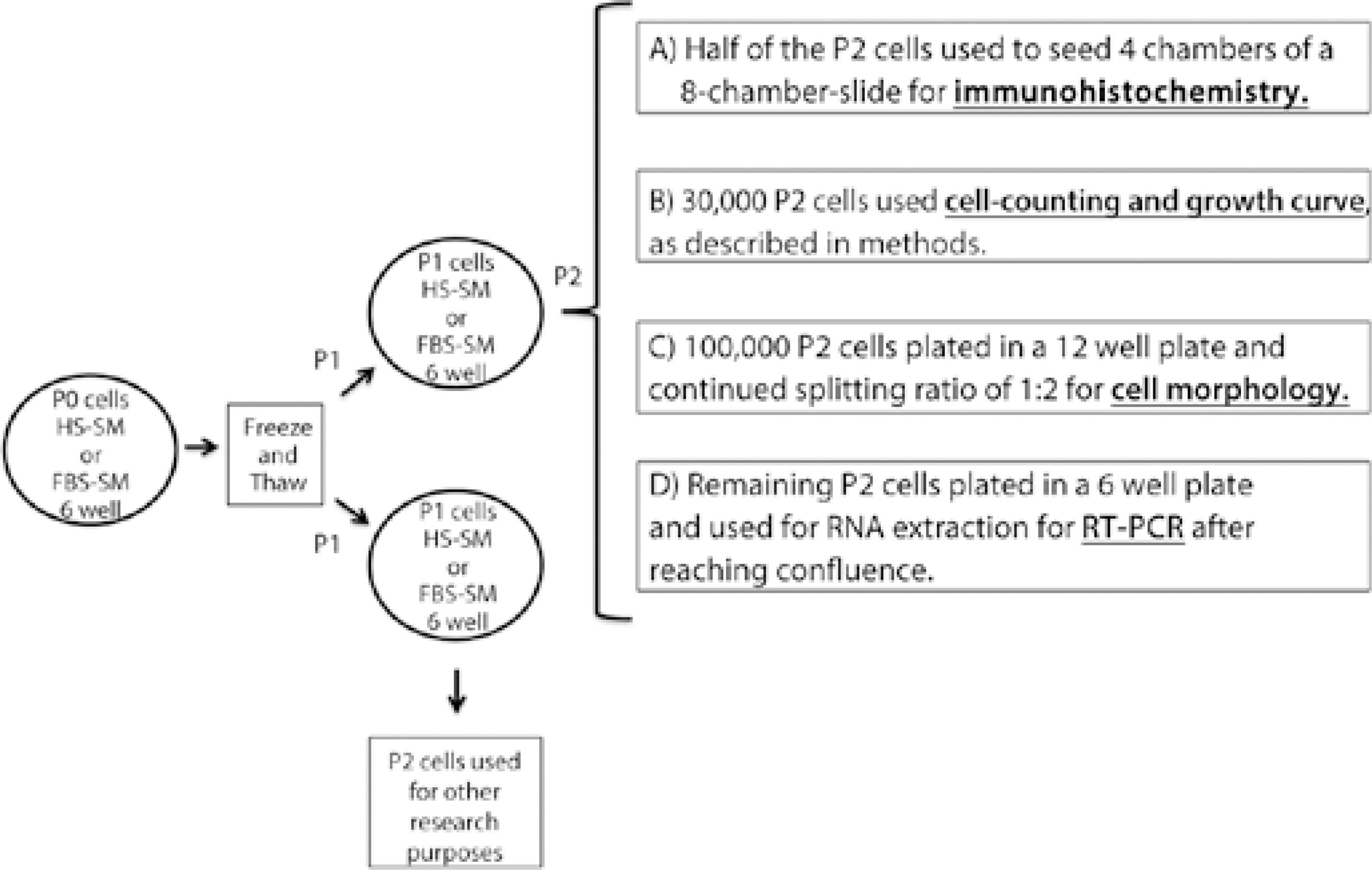
Real time-reverse transcriptase polymerase chain reaction
For each donor and medium, total RNA was extracted from confluent cultures in a single well of a 6-well plate at passage 2 (as described in Figure 1) using a commercially available kit (RNeasy Mini Kit; Qiagen, Valencia, CA) and stored at -80°C. The concentration and purity of the isolated RNA were determined using a NanoDrop instrument (Thermo Fisher Scientific Inc., Waltham, MA). Complementary DNA (cDNA) was obtained using the commercially available High Capacity RNA-to-cDNA™ kit (Life Technologies) and was stored at -20°C. Polymerase chain reaction (PCR) was conducted in a StepOnePlus Real Time PCR System (Life Technologies) with the prepared cDNA and commercially available TaqMan primer-probe sets (Life Technologies), targeting 1) ZO-1, tight junction protein 1 (TJP1); Na+/K+-ATPase, Na+/K+ transporting, alpha 1 polypeptide (ATP1A1); and GPC4. Gene expression was assessed by the comparative CT (ΔΔCT) method with β-actin as the normalizing gene. Assays were performed in triplicates for all samples.
Immunohistochemistry
For immunohistochemical analysis, cells were grown (using 16% serum-supplemented media) in chamber slides that were pre-coated with FNC Coating Mix, fixed in 4% paraformaldehyde for 15 min, and treated with 0.1% Triton X-100 for 10 min at room temperature. Non-specific binding was blocked by incubating cells in 10% donkey or goat serum for 1 h before incubation overnight at 4°C with the primary antibodies (all 1:100, ZO-1, Na+/K+-ATPase, and GPC4). Secondary staining was performed with secondary antibodies (1:500) for 1 h at room temperature, and cells were counterstained with Hoechst nuclear staining (1:2500). Cells were analyzed using a Zeiss LSM 510 Meta Confocal Microscope (Carl Zeiss, Berlin, Germany).
Growth curve
As shown in figure 1, 30,000 cells for each donor and medium were recovered from P2 and were used to seed 5,000 cells in five different wells of a 24-well plate (for counting on days 1, 2, 3, 5, and 7) and in one well of a 6-well plate (for counting on day 10). Thus, each well contained the same number of cells at different densities depending on the size of the well in which they were placed. At specified times, on plating (days 1, 2, 3, 5, 7, and 10), cells were trypsinized and counted using a microscope (IMT-2, Olympus, Tokyo, Japan) and a hemocytometer (Hausser Scientific, Horsham, PA). Three counts were performed in triplicates for each well, and the results were averaged to construct the growth curve.
Morphology assessment (phase contrast microscopy)
A phase-contrast microscope with a digital camera (ProgResC5, Jenoptik, Jena, Germany) was used to image confluent cells for each donor, media, and passage from P0 to P5 to document morphology data. The well was randomly placed under the microscope, and images were taken at 20× magnification from each passage.
Detachment time assessment
For each donor, the medium was aspirated, and cells were washed with HBSS, with the same amount of trypsin solution being added to all plates, at each passage. A standard amount of tapping was applied to the plate every 30 s to facilitate detachment. The time taken for complete cell detachment was counted for each of the media. If the detachment period amounted to 5 min and complete detachment was not achieved, cells were mechanically detached by adding media using a pipette.
RESULTS
Morphology
Typical endothelial-cell polygonal morphology was observed for cells grown in both FBS- and HS-SM at passage 1 (P1) for all three pairs of cell populations, and a typical spindle-shape transformation could be initially observed for both FBS- and HS-SM at P5 (Figure 2). No difference in morphology could be observed at P0, P1, P5 or at any other passages for cells grown in the two media before or after cryopreservation.
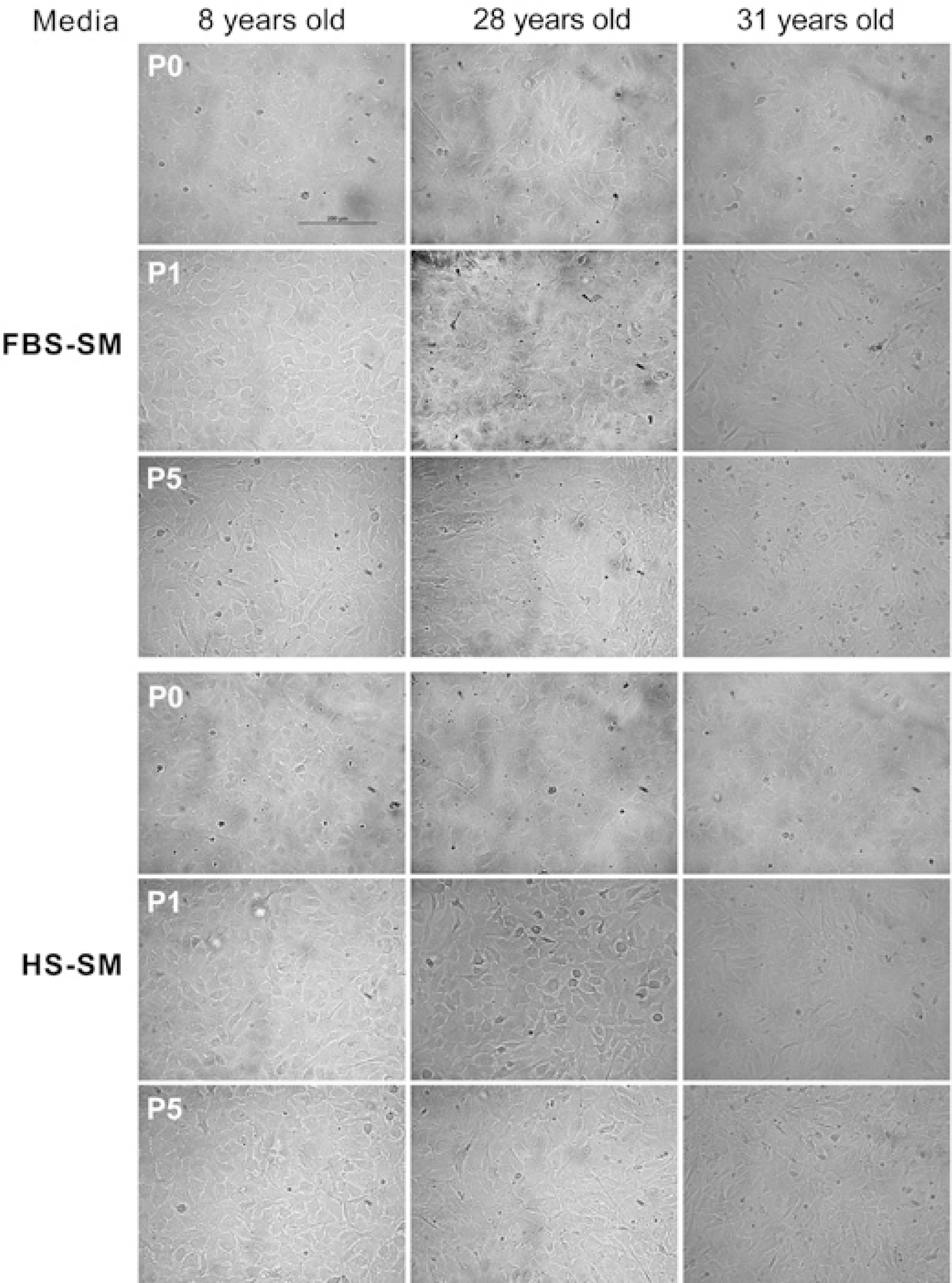
Figure 2 Phase-contrast images of confluent passage 0 (before cryopreservation) and passages 1 and 5 (after cryopreservation) HCECs cultured in fetal bovine serum-supplemented medium (FBS-SM) and human serum-supplemented medium (HS-SM). Similar cell morphologies were observed for the two media for the same cell populations and passages. All pictures were taken using 20× magnification. Scale bar=200 μm.
Growth characteristics
The time from initial plating (P0) to confluence was 7, 17, and 14 days for donor 1, 2, and 3, respectively, when grown in FBS-SM and 5, 15, and 14 days, respectively, when grown in HS. For subsequent passages, times were slightly different between cell populations when comparing FBS- and HS-SM for a given passage and cell population. Using growth curves, cell counts after thawing were similar for FBS- and HS-SM from days 1 to 10, with a slight trend toward higher cell counts in FBS-SM (Figure 3).
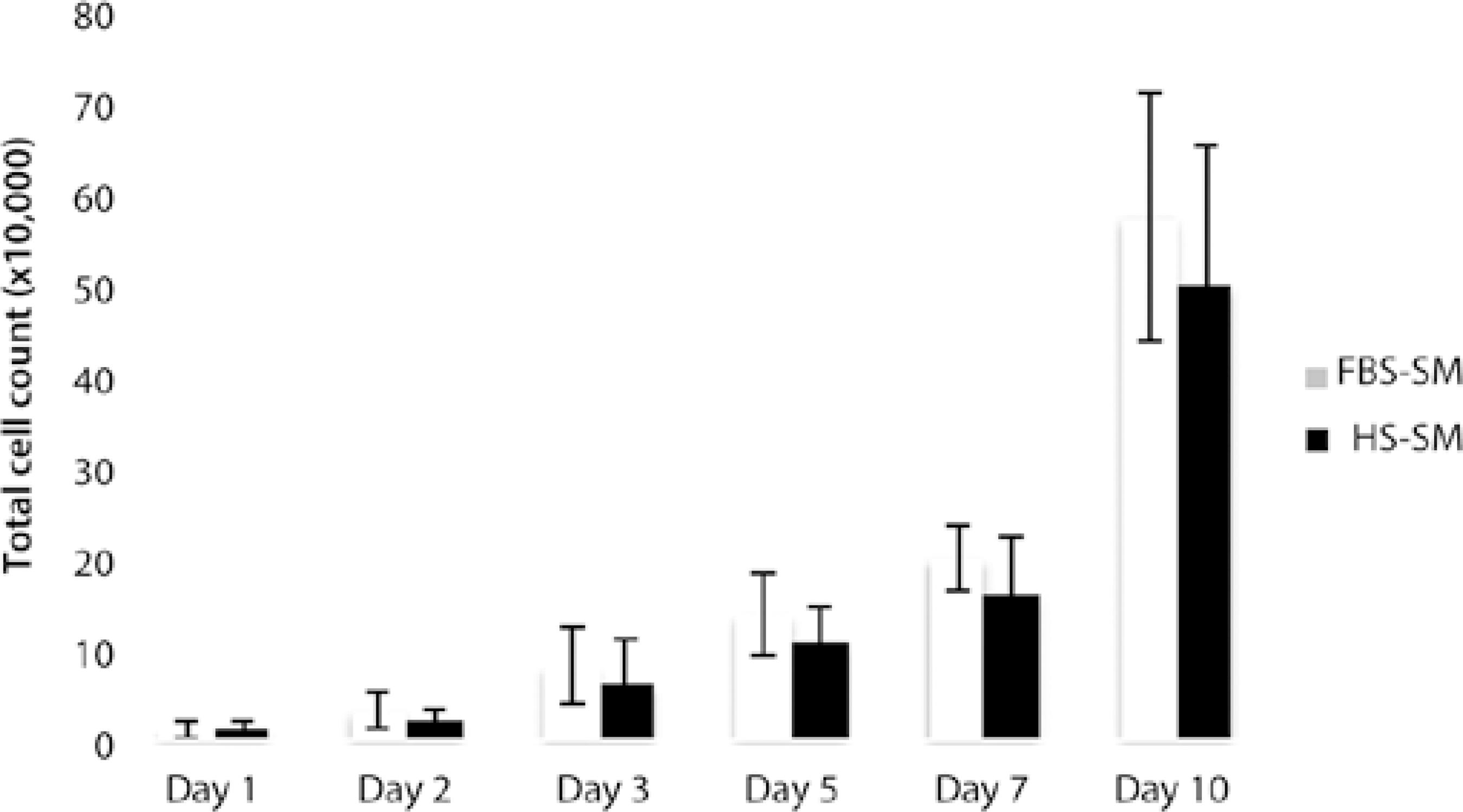
Figure 3 Ten-day growth curve comparing cells cultured in fetal bovine serum-supplemented medium and human serum-supplemented medium starting at passage 2. On days 1, 2, 3, 5, and 7 counts were obtained for cells seeded in a 24-well plate; at day 10, counts for cells seeded in a 6-well plate were obtained. Error bars represent standard deviations.
Immunofluorescence and real time-PCR
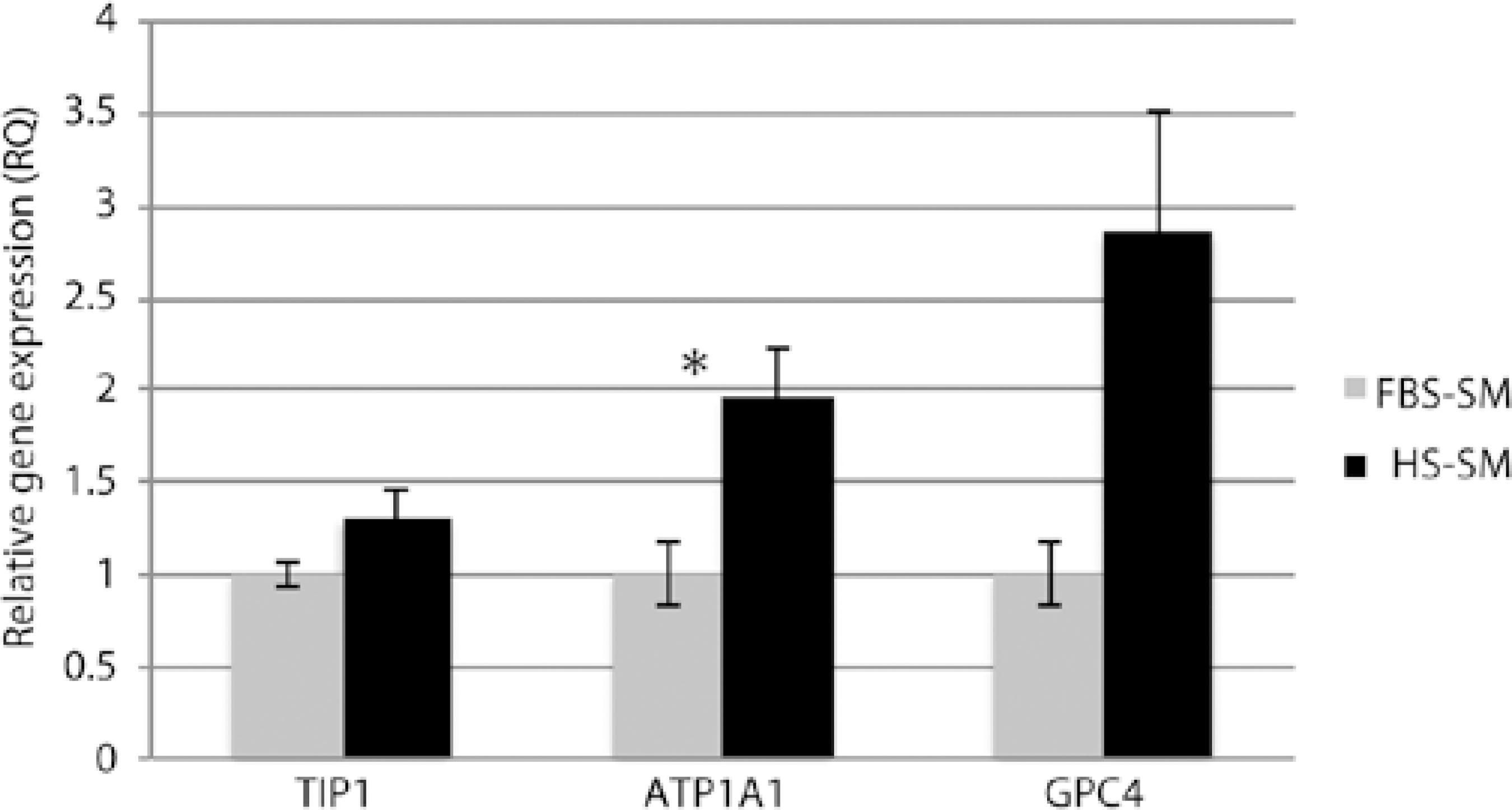
Cells grown in both FBS- and HS-SM demonstrated similar staining of functional (ZO-1 and Na+/K+-ATPase) and endothelial cell (GPC4) markers when assessed by immunohistochemistry (Figure 4). Negative controls using only secondary antibodies AlexaFluor 488 and 555 demonstrated no staining (Figure 4). When assessed by real time (RT)-PCR, gene expression of HCEC markers was higher in HS-SM than in FBS-SM: TJP1 (1.3-fold, P =0.083), ATP1A1 (two fold, P =0.031), and GPC4 (3-fold, P =0.064) (Figure 5).
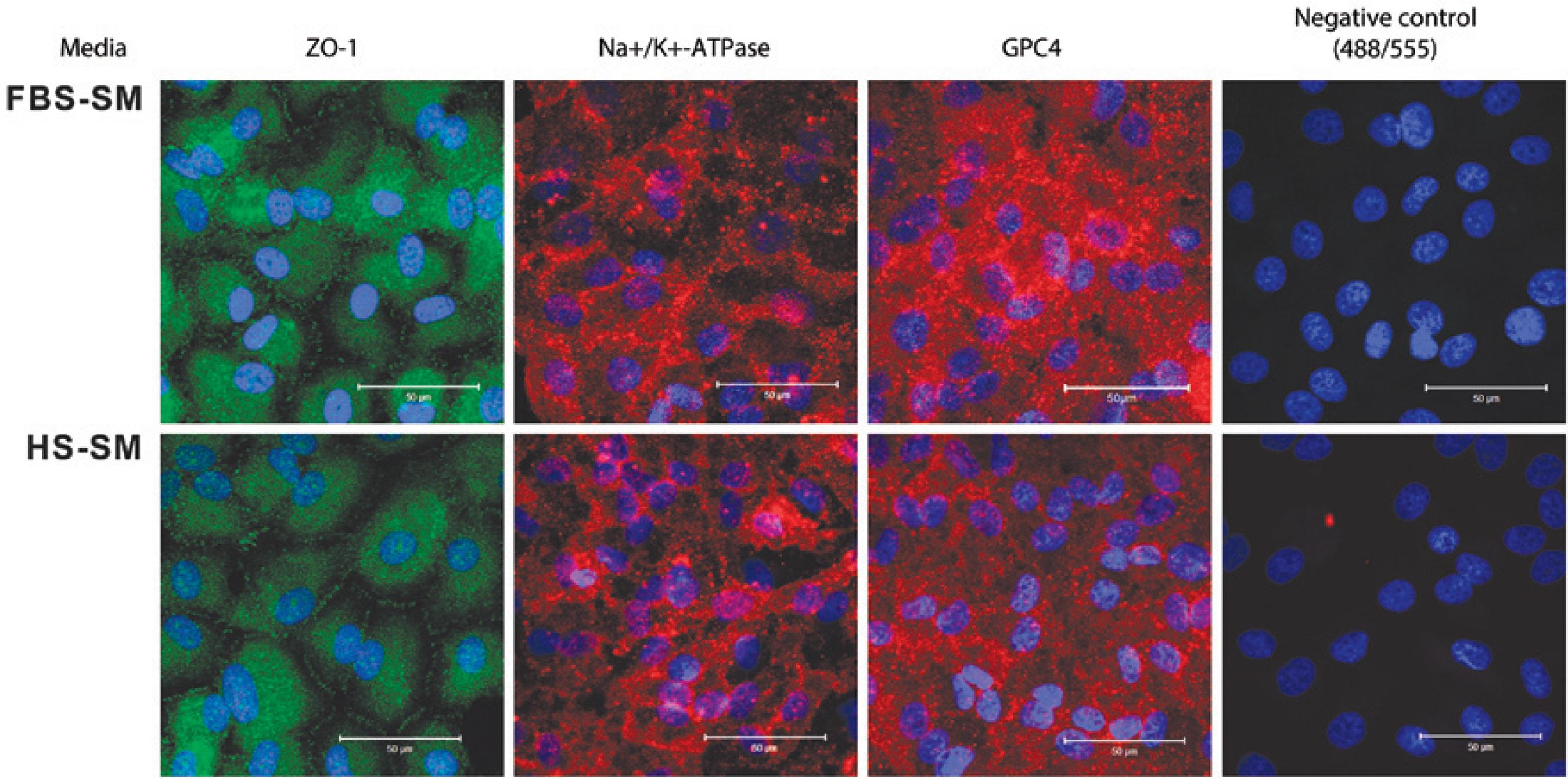
Figure 4 Immunostaining for zonula occludens-1 (ZO-1), sodium-potassium ATPase (Na+/K+-ATPase), and glypican 4 (GPC4) of confluent passage-2 HCECs cultured in media with fetal bovine serum (FBS-SM) or human serum (HS-SM). Staining for ZO-1 is from the 31-year-old donor; Na+/K+-ATPase and Negative Control [secondary antibodies AlexaFluor 488 (above) and 555 (below) only] from the 8-year-old donor, and GPC4 from the 31-year-old donor. Scale bars=50 micrometers.
Detachment time
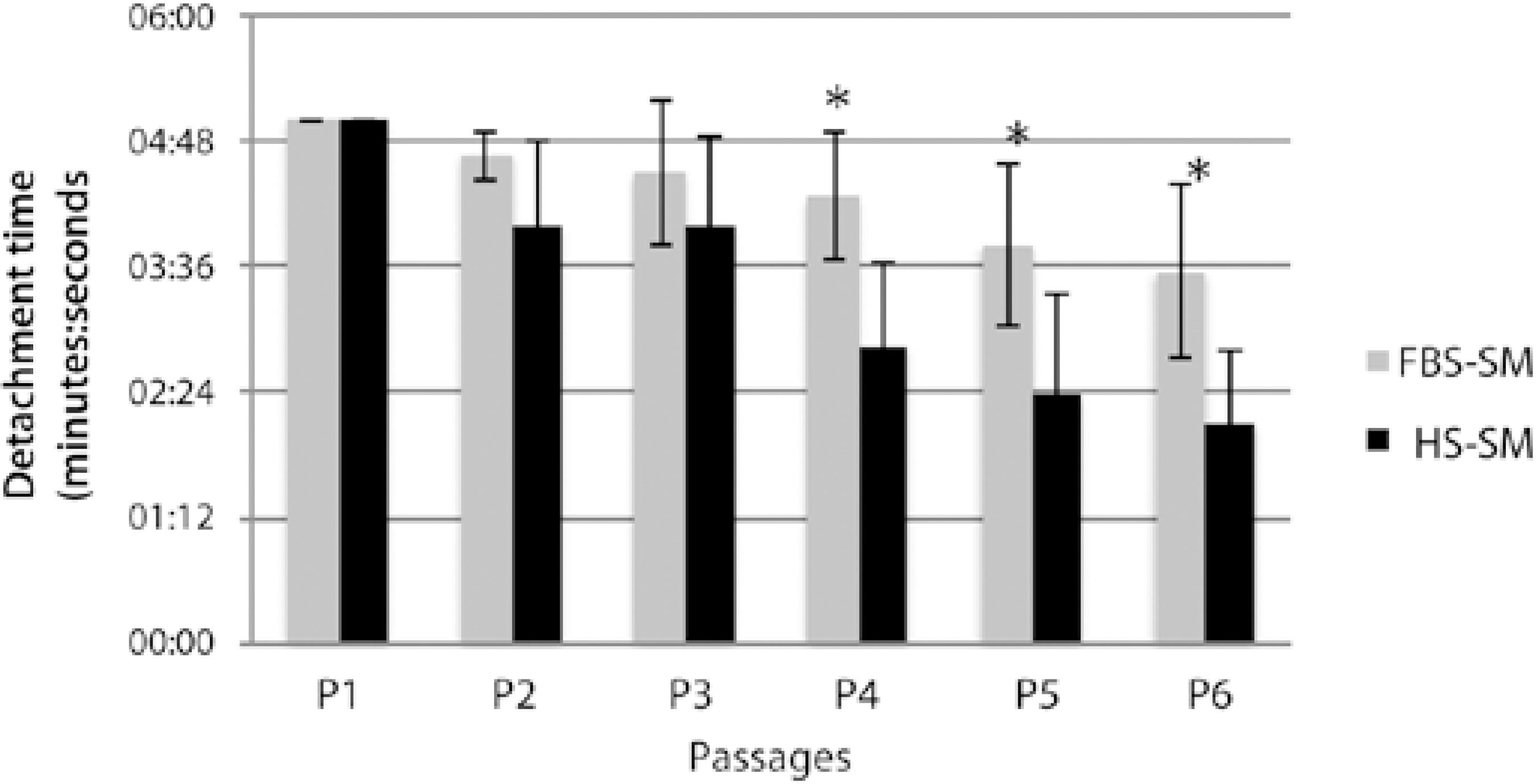
All cell populations from both the media reached the maximum time allowed for detachment from P0 to P1; thus, there was no difference between the populations at these passages. However, for P2 and for all subsequent passages, there was a trend toward longer detachment time with FBS-SM and lower passages (Figure 6), with statistical significance for passages 4 (P =0.0268), 5 (P =0.0279), and 6 (P= 0.0098).
DISCUSSION
The development in surgical treatment of corneal endothelial diseases, with progressive thinning of grafts and its related advantages(13,14), leads us to conclude that transplantation with cultured endothelial cells may represent the next advance in this field(2-9). Animal studies have demonstrated promising results(15,16). An important issue that may arise if this approach becomes feasible is cell maintenance and distribution to surgeons. Cryopreservation can mitigate these issues with some additional advantages, such as the long-term preservation of the endothelial cell source(5). We have recently demonstrated that human serum can be used as a supplement for HCEC culture, conferring the added advantage of a medium with a lower content of animal derivatives. In this study we demonstrated the ability of HCECs grown in HS-SM to be cryopreserved and maintain their characteristics by comparing them with cells grown in FBS-SM.
No evident difference in morphology of the cells could be observed when comparing FBS- and HS-SM cultures from P0 to P5 across all cell populations studied. Growth rates were also similar between the two media using passage-2 cultures, with a slight trend toward higher cell counts in FBS-SM. Both FBS- and HS-SM demonstrated similar immunostaining for functional (ZO-1 and Na+/K+-ATPase) and endothelial cell (GPC4) markers of passage-2 cultures. However, HCEC marker gene expression was higher in HS-SM when assessed by RT-PCR. The impact of this finding on the suitability of cryopreserved HCECs grown in HS-SM for use in cell therapy is unclear and warrants further study with an expanded number of cell populations and markers. Further testing should include physiological assays, such as the use of an Ussing chamber(17), to determine any differences in the pumping activity of HCECs grown in FBS- and HS-SM with and without cryopreservation.
Unpublished observations during our previous study(9) have demonstrated that cells grown in HS-SM detach faster than those grown in FBS-SM. Similarly, in this study, we observed a tendency of longer detachment time with FBS-SM and lower passages. Moreover, we observed that when cells are plated in glass chamber slides with HS-SM (8% as standard), they can spontaneously detach even with the use of a cell-attachment reagent, which does not occur when 16% HS-SM or 8% FBS-SM is used. Further studies are warranted to establish the cause of this finding and its potential clinical implications.
In summary, we demonstrated that on cryopreservation, HS-SM was similar to FBS-SM for HCEC culture as assessed by comparison of cell morphology, proliferation, and protein expression, although marker gene expression by RT-PCR was higher in HS-SM-based cultures than in FBS-SM-based cultures. Detachment time was longer with FBS-SM and lower passages. To establish the relevance of our findings with regard to the potential clinical use of cryopreserved HCECs cultured in HS-SM, follow-up studies should be conducted that which would further contribute to the progress toward the ultimate goal of endothelial cell therapy.

