INTRODUCTION
The term pachychoroid neovasculopathy (PN) was first coined by Freund(1) to describe a disease characterized by a form of type 1 (subretinal pigment epithelium) neovascularization involving dilated choroidal vessels in areas of increased choroidal thickness. Recently, it was posited that PN(1) should be grouped with macular diseases, including central serous chorioretinopathy(2) and pachychoroid pigment epitheliopathy(3), in which choroidal thickening of the macular region is demonstrated by enhanced depth imaging (EDI)-optical coherence tomography (OCT). In the first published paper regarding PN, Pang and Freund(1) described the occurrence of PN in three white female patients. Herein, we report the case of a male patient diagnosed with PN and provide descriptions of our subsequent multimodal evaluation of this patient.
CASE REPORT
We report the case of a 58-year-old white male who presented three years previously with impaired visual acuity affecting the left eye. The visual acuity was 20/25 in the right eye and 20/200 in the left eye. Ocular fundus examination revealed reduced fundus tessellation in the right eye (Figure 1 A). A semitranslucent epiretinal membrane caused macular distortion. Membrane contraction displaced the paramacular vessels toward the horizontal raphe in the left eye (Figure 1 A). Fluorescein angiography (FA) of the right eye demonstrated no window defects at any location. The left eye displayed staining in the vascular area of the epiretinal membrane (Figure 1 B). Spectral domain (SD)-OCT revealed retinal pigment epithelium abnormalities on the right eye and an epiretinal membrane with wrinkling of the inner retina of the left eye (Figure 1 C).
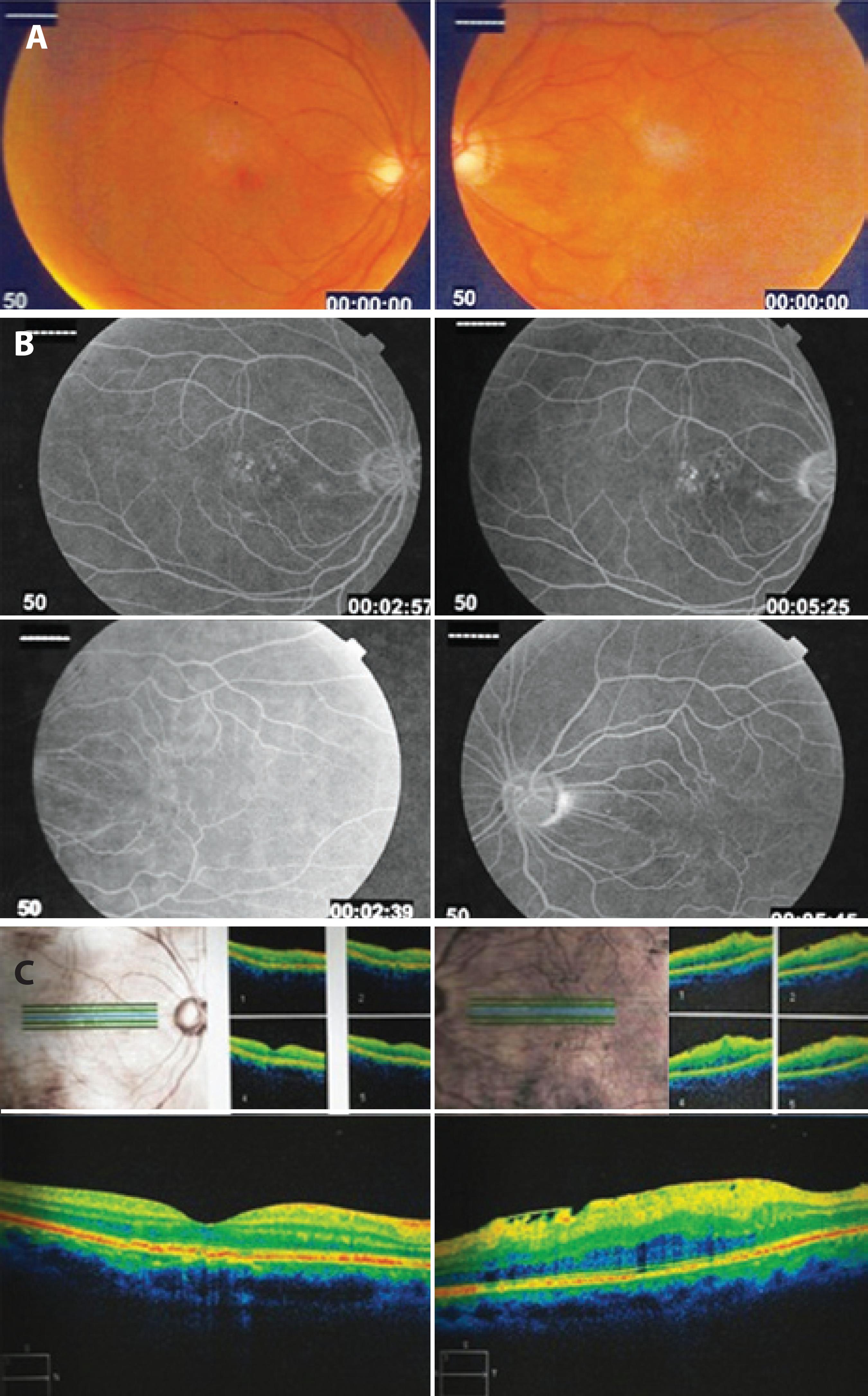
Figure 1 A) Fundus tessellation in the right eye and an epiretinal membrane causing distortion of the macula in the left eye. B) Window defects in the right eye. The epiretinal membrane displaced the paramacular vessels toward the horizontal raphe in the left eye. C) SD-OCT images demonstrating RPE abnormalities in the right eye and epiretinal membrane wrinkling of the inner retina in the left eye.
Two years after epiretinal membrane surgery on the left eye, the patient presented with a decrease in visual acuity of the right eye to 20/40; however, the visual acuity in the left eye had improved to 20/100. Multimodal evaluation was performed. Color photographs of the right eye revealed further reduction of fundus tessellation (Figure 2 A). In contrast to the examination at the initial presentation demonstrating displacement of paramacular vessels toward the horizontal raphe, FA demonstrated poorly demarcated leakage in the right eye and regular retinal capillary calibers in the left eye (Figure 2 B). SD-OCT of the right eye revealed small pigment epithelial detachments and subretinal fluid. In the left eye, the epiretinal membrane and wrinkling of the inner retina had persisted from the initial presentation (Figure 3 A). By EDI-OCT, subchoroidal thicknesses in the affected right eye and normal left eye were 247 μm and 165 μm, respectively (Figure 3 B).
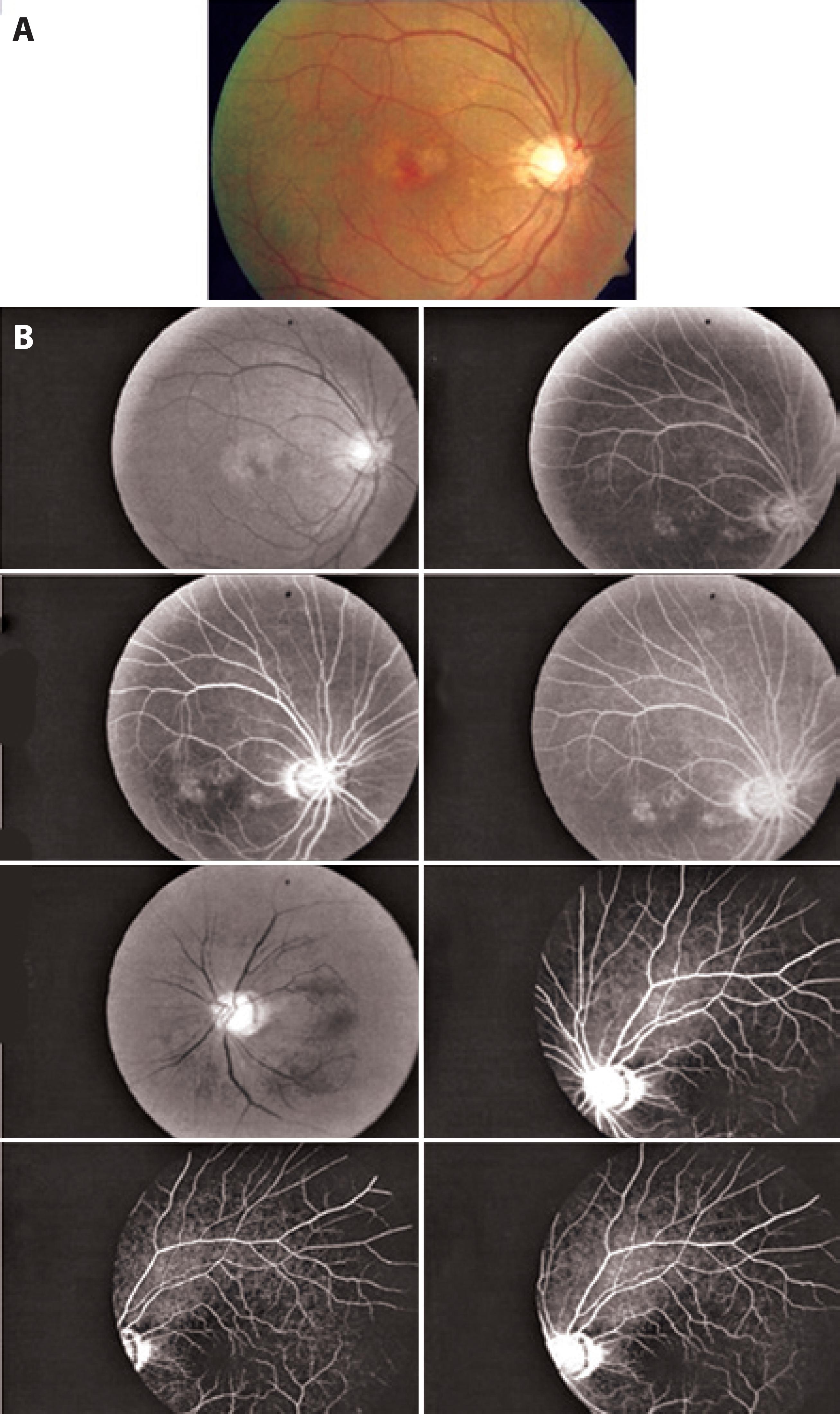
Figure 2 Progression of fundus tessellation two years later. B) FA two years later demonstrating poorly demarcated leakage in the right eye and regularity of retinal capillaries in the left eye.
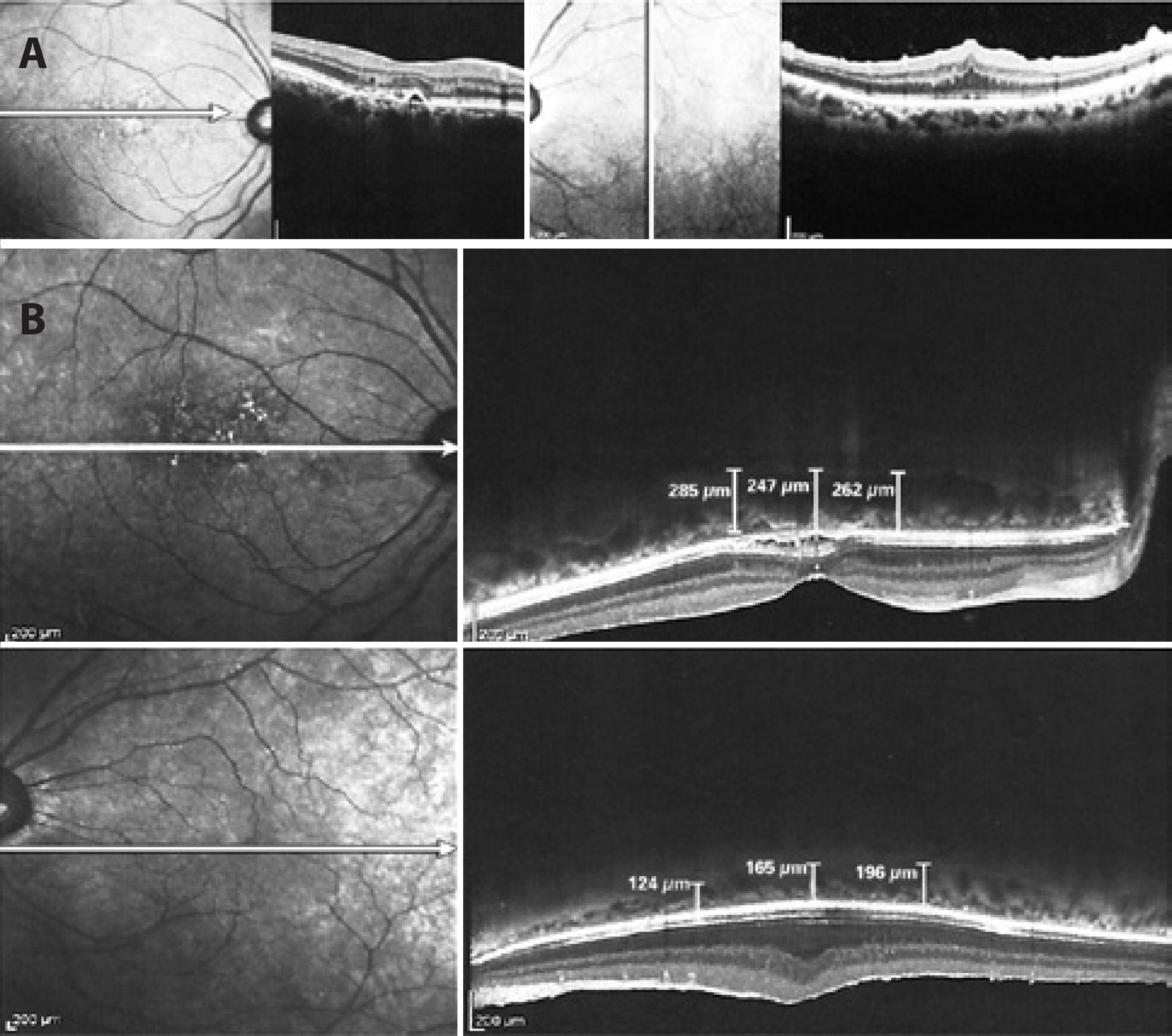
Figure 3 A) SD-OCT images demonstrating a small PED and subretinal fluid in the right eye and persistence of the epiretinal membrane and wrinkling of the inner retina in the left eye. B) EDI-OCT measurement of subfoveal choroid thickness in the affected right eye and normal left eye.
Fundus autofluorescence was used to detect hyperfluorescent regions in the right eye. No abnormalities were observed in the left eye (Figure 4 A). Indocyanine green angiographic analysis of the area of hyperfluorescence in the right eye was consistent with leakage from a Type 1 occult choroidal neovascularization (CNV). No abnormalities consistent with CNV were observed in the left eye (Figure 4 b).
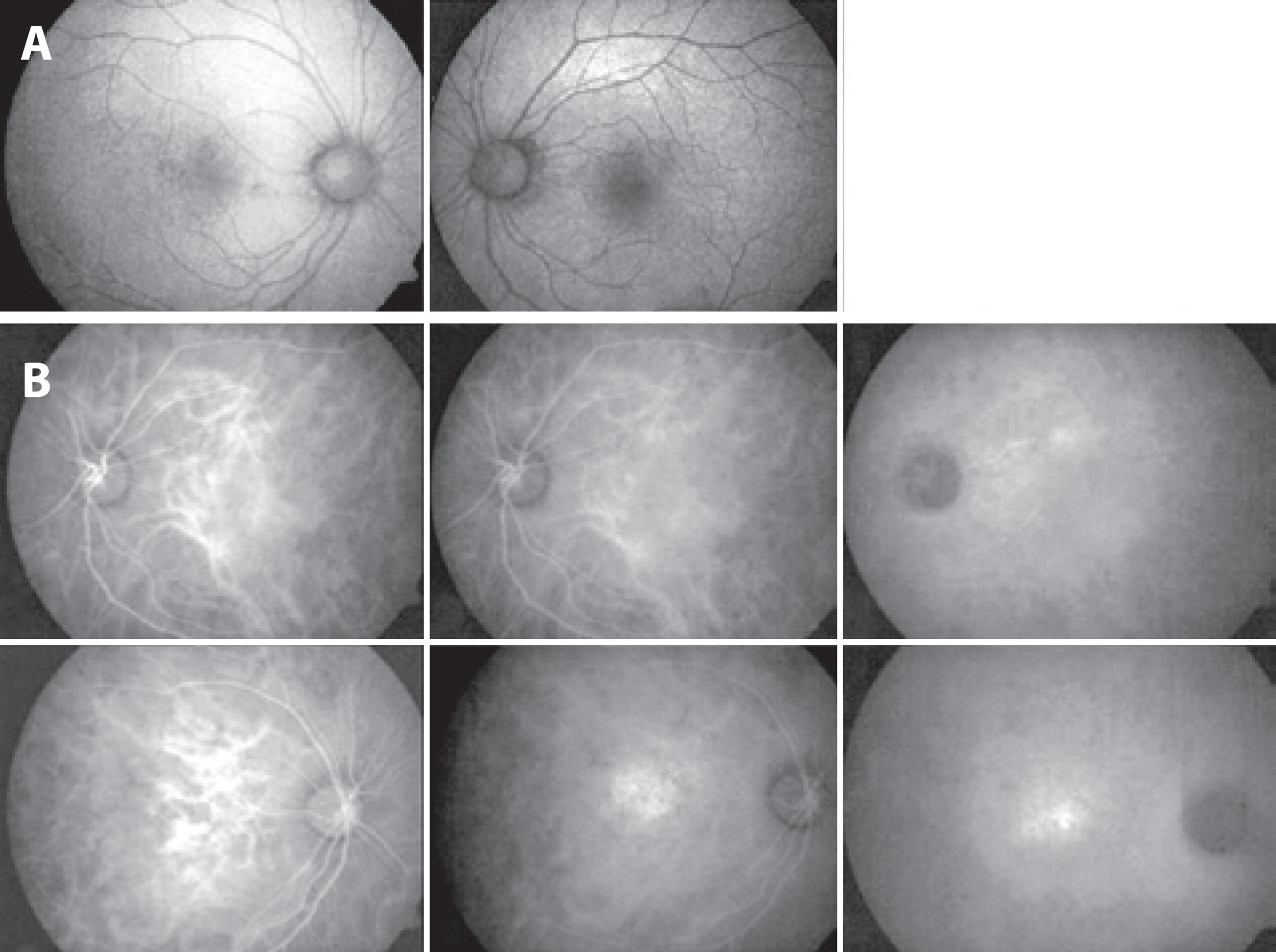
Figure 4 A) FAF demonstrating hypoautofluorescent regions in the right eye and no abnormalities on the left eye. B) Indocyanine green angiography revealed a region of hypofluorescence consistent with leakage from a type 1 occult CNV. No abnormalities consistent with CNV were observed in the left eye.
DISCUSSION
PN was originally reported in three white female patients(1). In this report, we describe a case of PN in a male patient. Thus, PN does not appear to be gender-specific and may occur in white male populations. The original paper(1) reported a mean subfoveal choroidal thickness in affected eyes of 310 μm with a range of 244 μm-407 μm. In our case study, a subfoveal choroidal thickness of 247 μm was recorded in the affected eye; thus, our results corroborate the EDI-OCT findings of the original report(1).
In the original paper, polypoidal vascular lesions were observed in two of the three patients but were not observed in the patient described in this report at any point during diagnosis or treatment. Polypoidal choroidal vasculopathy, primarily a choroidal pathology, may occur as a result of long-standing Type 1 CNV in central serous chorioretinopathy (CSCR)(4) or age-related macular degeneration (AMD)(4). The patient in this report had no history of AMD or preexisting conditions that may predispose to Type 1 CNV. An inherited cause(5) has been posited to underlie pachychoroid retinal diseases as well as CSCR(6).
In conclusion, we present a clinical case of PN in a white male patient in contrast to the original description of PN in three white female patients.

