Arq. Bras. Oftalmol. 2012; 75 (3): 10.1590/S0004-27492012000300013
Total: 3167
Alexandre Tagliari Cestari1; Juliana Maria Ferraz Sallum2; Marina Lourenço De Conti2; Thiago Iorio Tagliari3; Marcello Novoa Colombo Barboza
DOI: 10.1590/S0004-27492012000300013
RESUMO
A retinose pigmentada constitui um grupo de doenças causadas por alterações genéticas que levam à degeneração progressiva dos fotorreceptores, principalmente bastonetes. Em geral, tem apresentação bilateral. Este estudo é um relato de caso de uma paciente com acometimento unilateral da retina, de características semelhantes às da retinose pigmentada, com história de trauma ocular antigo. Descrevem-se sua história clínica e achados oftalmológicos.
Descritores: Retinite pigmentosa; Traumatismos oculares; Distrofias retinianas; Relatos de casos
ABSTRACT
Retinitis pigmentosa is a group of diseases caused by genetic changes that lead to progressive degeneration of photoreceptors, rods mainly. In general, it has bilateral presentation. This study is a case report of a patient with unilateral involvement of the retina, similar to the characteristics of retinitis pigmentosa, and an old ocular trauma history. It describes her history and ophthalmologic findings.
Keywords: Retinitis pigmentosa; Eye injuries; Retinal dystrophies; Case reports
INTRODUÇÃO
O termo retinose pigmentada (RP) refere-se a um grupo de doenças genéticas que levam à degeneração progressiva dos fotorreceptores, principalmente bastonetes. Na maioria das vezes, a RP apresenta-se de forma bilateral e simétrica, e os casos mais comuns aparecem no adolescente ou adulto jovem. O fundo de olho é caracterizado por estreitamento de vasos sanguíneos e presença de pigmento retiniano, em formato de espículas ósseas. O comprometimento ocorre principalmente na média periferia e periferia, podendo acometer visão central e pólo posterior(1,2).
A evolução típica consiste em diminuição da visão noturna e de campo visual lateral. O eletrorretinograma evidencia diminuição progressiva da função celular(1), e o campo visual pode revelar escotomas, com possível evolução para campo visual tubular.
Outras doenças genéticas podem causar alterações retinianas semelhantes àquelas da RP (fenocópias). As alterações podem ser ainda resultado de infecção (sífilis, toxoplasmose, rubéola, varicela, sarampo, citomegalovirose), deficiência de vitamina A, efeito de medicamento (cloroquina, clorpromazina, tioridazina), neoplasias (Retinopatia associada a câncer - CAR), eventos vasculares, degeneração senil da retina e trauma ocular(3).
A retinose pigmentada unilateral é uma rara distrofia da retina, cujo diagnóstico inclui achados morfofuncionais compatíveis com RP no olho afetado e olho contralateral normal em aspecto anatômico, eletrorretinograma e campo visual. Deve-se acompanhar o paciente por período mínimo de 5 anos para se excluir desenvolvimento de alterações no olho não afetado(4). Causas inflamatórias, traumáticas e tóxicas também devem ser excluídas(5).A existência da RP unilateral como uma doença genética permanece não esclarecida(6,7).
RELATO DO CASO
L.H.S.F., sexo feminino, parda, 54 anos. Procurou atendimento oftalmológico na cidade de Catanduva - SP, em outubro/2010, referindo embaçamento visual para longe e perto.
Apresentava história de trauma em olho direito há 25 anos, sem acompanhamento oftalmológico desde então. Negou cegueira, doenças genéticas ou oftalmológicas na família. Boa saúde geral.
O exame oftalmológico constatou acuidade visual 20/20 em ambos os olhos (AO) com melhor correção, OD +1,00 DE // -0,50 DC a 145º e OE +1,75 DE // -0,25 DC a 155º. Adição +2,50 DE em AO.
Biomicroscopia sem anormalidades.
Tonometria 12 mmHg em AO.
Na avaliação fundoscópica observou-se OD com disco óptico de coloração normal e escavação fisiológica. Mácula preservada. Alteração de pigmentação do epitélio retiniano periférico em 360º em região extra-arcadas vasculares, com espículas e aspecto característico de retinose pigmentada. Concentricamente, dentro da região de arcadas, delimitou-se segunda área de coloração diferente da coloração macular, correspondendo a um anel hiperautofluorescente relacionado à progressão da retinose. Vasos afinados e opacidade vítrea móvel linear na projeção do disco óptico (Figuras 1 e 2). OE dentro da normalidade.
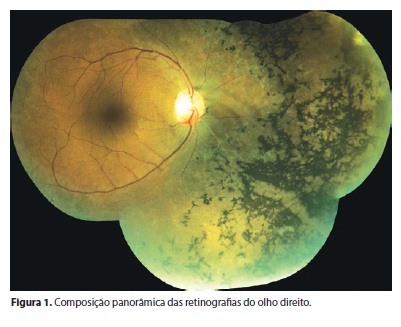
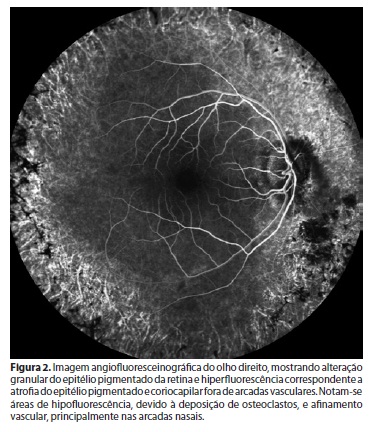
Na angiofluoresceínografia do OE na região extra-arcada nasal e nasal superior, notou-se diferença do padrão da coriocapilar - variações dentro da normalidade (Figuras 3 e 4).
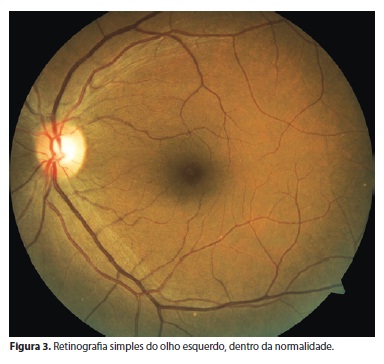
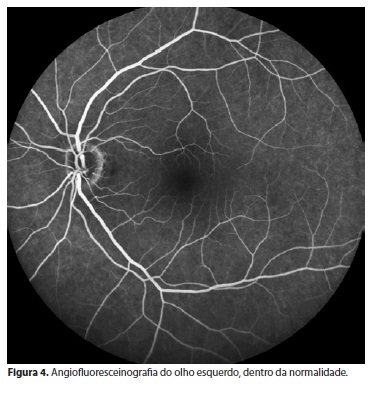
Na campimetria do OD, há constrição concêntrica de campo periférico e preservação do campo central de 20º. OE sem alterações.
A paciente foi submetida à eletrorretinografia padrão de campo total (ERG - ISCEV Padrão). Em ERG escotópico, verificou-se OD com resposta residual nos estímulos dos bastonetes, flash padrão e de alta intensidade. OE dentro da normalidade.
No exame de tomografia de coerência óptica, a retina apresentou características normais; na área mais temporal do corte no OD, nota-se afinamento da retina e ausência da camada de fotorreceptores, compatível com a transição da área de anel de hiperautofluorescência.
DISCUSSÃO
No caso apresentado, o acometimento retiniano não ocorre bilateralmente, devendo-se considerar formas de retinose pigmentada unilateral, mesmo que raras.
A paciente apresentava sorologia negativa para sífilis e rubéola, não possuía idade compatível com degeneração senil, nem histórico de eventos vasculares, neoplasias ou utilização de medicamentos associados a alterações da retina.
A história de trauma antigo, há 25 anos, permite a hipótese de retinose pigmentada secundária a trauma, dados o acometimento unilateral (no olho traumatizado) e a aparência não evolutiva da doença.
Não foi realizado seguimento oftalmológico após o trauma, o que dificulta o estabelecimento de relação precisa entre as alterações retinianas e o evento traumático.
A retinose pigmentada de origem genética apresenta padrão de herança autossômico dominante, autossômico recessivo ou ligado ao X. A proporção de casos isolados é elevada, cerca de 41%(7).
A forma unilateral da doença é de difícil explicação genética, mas algumas hipóteses de mecanismos podem ser levantadas. No mosaicismo, o genoma das células de um olho poderia conter mutações que não estariam presentes no outro olho. Na forma ligada ao X, mulheres portadoras do gene podem apresentar sinais menores da doença com padrão de pigmentação irregular devido à inativação do cromossomo X em cada célula. As mulheres portadoras são, em geral, míopes - a paciente relatada é hipermetrope(2). Outro fenômeno conhecido é o salto de gerações, presente em algumas formas da doença dominante. Os portadores de mutação neste gene, que não manifestam a doença, possivelmente têm um fator de proteção - um polimorfismo genético, por exemplo. Na entidade genética a atrofia coriorretiniana pigmentada paravenosa é descrita manifestação unilateral e está bem caracterizada mutação genômica no gene CRB1(3).
Mecanismos genéticos ainda não bem estabelecidos poderiam causar expressões diferentes entre os dois olhos(8,9).
A discussão sobre diferentes hipóteses diagnósticas e condutas a serem tomadas é fundamental para o esclarecimento do prognóstico do paciente em casos como o apresentado, direcionando adequadamente seu acompanhamento.
A retinose pigmentada unilateral de origem genética é uma entidade clínica bastante rara, que não pode ser confirmada no caso apresentado, uma vez que o estudo genético da paciente não pode ser realizado. É mais provável que se trate de uma fenocópia - uma pseudoretinose pigmentada unilateral, pós-trauma.
Um diagnóstico de retinose pigmentada, definido sem atenção à história clínica, levaria à previsão de perda visual progressiva no olho afetado e provável acometimento também do olho não afetado. Tal diagnóstico geraria implicações médicas e emocionais à paciente e não levantaria a discussão sobre RP secundária a trauma, condição sem caráter evolutivo e de melhor prognóstico, ainda pouco descrita.
REFERÊNCIAS
1. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa; Lancet. 2006;368(9549):1795-809.
2. Krill AE. Krill's hereditary retinal and choroidal diseases. Maryland: Harper & Row; 1977.
3. Ryan SJ. Retina. Basic science and Iiherited retinal disease. St. Louis: Mosby; 2001.
4. François J, Verriest G. Rétinopathie pigmentaire unilateral. Ophthalmologica. 1952; 124(2):65-88.
5. Carr RE, Siegel IM. Unilateral retinitis pigmentosa. Arch Ophthalmol. 1973;90(1):21-6.
6. Farber DB, Heckenlively JR, Sparkes RS, Bateman JB. Molecular genetics of retinitis pigmentosa. West J Med. 1991;155(4):388-99. Comment in: West J Med. 1001;155(4):423-4.
7. Unonius N, Farah ME, Sallum JMF. Classificação diagnóstica dos portadores de doenças degenerativas de retina, integrantes dos grupos Retina São Paulo e Retina Vale do Paraíba. Arq Bras Oftalmol. 2003;66(4):443-8.
8. Kolb H, Galloway NR. Three cases of unilateral pigmentary degeneration. Br J Ophthalmol. 1964;48:471-9.
9. Farrell DF. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin Ophthalmol. 2009;3:263-70.
Endereço para correspondência:
Alexandre Tagliari Cestari.
Avenida Deputado Orlando Zancaner, 555
Catanduva (SP) - 15801-120 - Brasil
E-mail: [email protected]
Submetido para publicação: 4 de outubro de 2011
Aceito para publicação: 9 de dezembro de 2011
Trabalho realizado no Departamento de Oftalmologia, Universidade Federal de São Paulo - UNIFESP.
Financiamento: Não houve financiamento para este trabalho.
Divulgação de potenciais conflitos de interesse: A.T.Cestari, Não; J.M.F.Sallum, Não; M.L.De Conti, Não; T.I.Tagliari, Não; M.N.C.Barboza, Não.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.