Arq. Bras. Oftalmol. 2011; 74 (6): 10.1590/S0004-27492011000600015
Total: 1811
Alexandre Achille Grandinetti1; Ezequiel Portella1; Jayme Arana2; Natasha Tatiana Vieira Iskorostenski3
DOI: 10.1590/S0004-27492011000600015
ABSTRACT
The authors describe the clinical characteristics of a 14-year-old female patient with ophthalmoscopic and angiographic aspects of Stargardt's disease in both eyes who developed subretinal fibrosis and localized retinal pigment epithelial hyperplasia in the posterior pole of the right eye 6 months after a mild blunt trauma to the right globe. The left fundus remains with the typical presentation of Stargardt's disease. Our findings support the theory that ocular trauma occurring in patients with Stargardt's disease can precipitate a sequence of events leading to the development of subretinal fibrosis and significant visual loss.
Keywords: Retinal diseases; Fibrosis; Retinal pigment epithelium; Macular degeneration; Visual acuity; Case reports
RESUMO
Os autores descrevem as características clínicas de uma paciente de 14 anos com aspectos na oftalmocospia e angiofluoresceinografia de doença de Stargardt em ambos olhos que desenvolveu fibrose sub-retiniana e hiperplasia localizada do epitélio pigmentado da retina no polo posterior do olho direito seis meses após um trauma contuso leve neste globo. A fundoscopia do olho esquerdo permanence com a apresentação típica de doença de Stargardt. Nossos achados suportam a teoria de que o trauma ocular em pacientes com doença de Stargardt pode precipitar uma sequência de eventos que pode levar ao desenvolvimento de fibrose sub-retiniana e acentuada baixa da acuidade visual.
Descritores: Doenças retinianas; Fibrose; Epitélio pigmentado da retina; Degeneração macular; Acuidade visual; Relato de casos
CASE REPORTS RELATOS DE CASOS
Subretinal fibrosis in stargardt's disease: case report
Fibrose sub-retiniana na doença de Stargardt: relato de caso
Alexandre Achille GrandinettiI; Ezequiel PortellaI; Jayme AranaII; Natasha Tatiana Vieira IskorostenskiIII
IPhysician, Setor de Retina e Vítreo do Hospital das Clínicas da Universidade Federal do Paraná - UFPR - Curitiba (PR), Brazil
IIPhysician, Departamento de Oftalmologia da Faculdade Evangélica do Paraná - FEPAR - Curitiba (PR), Brazil
IIIPhysician, Universidade Federal do Paraná - UFPR - Curitiba (PR), Brazil
ABSTRACT
The authors describe the clinical characteristics of a 14-year-old female patient with ophthalmoscopic and angiographic aspects of Stargardt's disease in both eyes who developed subretinal fibrosis and localized retinal pigment epithelial hyperplasia in the posterior pole of the right eye 6 months after a mild blunt trauma to the right globe. The left fundus remains with the typical presentation of Stargardt's disease. Our findings support the theory that ocular trauma occurring in patients with Stargardt's disease can precipitate a sequence of events leading to the development of subretinal fibrosis and significant visual loss.
Keywords: Retinal diseases; Fibrosis; Retinal pigment epithelium; Macular degeneration; Visual acuity; Case reports
RESUMO
Os autores descrevem as características clínicas de uma paciente de 14 anos com aspectos na oftalmocospia e angiofluoresceinografia de doença de Stargardt em ambos olhos que desenvolveu fibrose sub-retiniana e hiperplasia localizada do epitélio pigmentado da retina no polo posterior do olho direito seis meses após um trauma contuso leve neste globo. A fundoscopia do olho esquerdo permanence com a apresentação típica de doença de Stargardt. Nossos achados suportam a teoria de que o trauma ocular em pacientes com doença de Stargardt pode precipitar uma sequência de eventos que pode levar ao desenvolvimento de fibrose sub-retiniana e acentuada baixa da acuidade visual.
Descritores: Doenças retinianas; Fibrose; Epitélio pigmentado da retina; Degeneração macular; Acuidade visual; Relato de casos
INTRODUCTION
Stargardt's disease (STGD) is a bilateral, symmetrical and progressive macular dystrophy that usually starts between the ages of 6 and 20 years and rapidly leads to loss of central vision(1). It is the most common form of hereditary macular dystrophy and accounts for 7% of all retinal dystrophies(2-4).
Genetically, STGD is a heterogeneous disorder that is usually inherited as an autosomal recessive disorder but rarely can present as an autosomal dominant trait with a later onset of clinical symptoms(1,5). The fundus findings may be limited to an atrophic maculopathy, which may have a bull's eye appearance. The macular changes may be associated with yellow pisciform flecks in the posterior pole and mid-periphery that can increase in size and number and eventually disappear. Atypical fundus findings have been described, including pallor of the optic disc, attenuated retinal vessels, retinal pigmentation in the form of bone spicule, cicatricial chorioretinitis, retinal pigment hyperplasia, subretinal neovascularization and subretinal fibrosis(1,6).
Previous histopathological studies using a noninvasive fundus spectrophotometer and immunocytochemical markers have documented structural changes in the retinal pigment epithelium (RPE) and an abnormal accumulation of lipofuscin, which is thought to be responsible for the retinopathy seen in Stargardt's disease(1,6,7). An angiographic characteristic sign is the dark choroid, also known as choroidal silence, that probably results from the accumulation of abnormal material in the RPE, presumed to be lipofuscin(1).
We report the presence of massive subretinal fibrosis in one eye of a patient with Stargardt's disease with a history of blunt ocular trauma. Our findings support the theory that ocular trauma occurring in patients with STGD can precipitate a sequence of events leading to the development of subretinal fibrosis and significant visual loss(1,6).
CASE REPORT
A 14-year-old girl was referred due to decreased visual acuity on the right eye. Her first eye exam was performed 3 years prior, and showed a visual acuity of 20/25 on both eyes (-1,00 sphere OU) with normal biomicroscopy and intraocular pressure. A dilated fundus exam was not performed at that time.
She complained of progressive decrease of visual acuity on her right eye, with a history of being struck by a volley ball in the head four months prior. Her medical and family histories were negative.
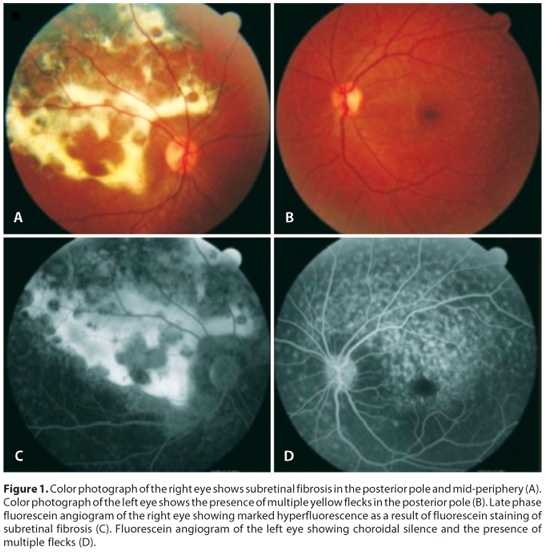
Visual acuity was 20/200 in the right eye and 20/30 on the left eye (-1,00 -2,00 180º OD, -1,25 -0,25 30º OS). The external examination was normal on both eyes, as well as the biomicroscopic examination of the anterior segment. The intraocular pressure was measured using a Goldmann tonometer, being 12 mmHg in the right eye and 13 mmHg in the left eye. Gonioscopy was performed on both eyes using a three-mirror lens and found no abnormalities. The fundus examination of the right eye showed yellow flecks in the posterior pole and mid-periphery with extensive subretinal fibrosis and diffuse areas of subretinal hyperpigmentation consistent with reactive hyperplasia of the RPE. The left fundus showed yellow flecks in the posterior pole and minimal RPE pigment mottling within the macula.
Fluorescein angiography showed choroidal silence with focal areas of hyperfluorescence corresponding to the flecks on both eyes (Figure 1). The late phase angiogram of the right eye showed marked hyperfluorescence in the posterior pole and superior mid-periphery secondary to severe RPE atrophy with some areas of blockade of fluorescence secondary to reactive hyperplasia of the RPE. The areas of subretinal fibrosis showed marked hyperfluorescence as a result of fluorescein staining.
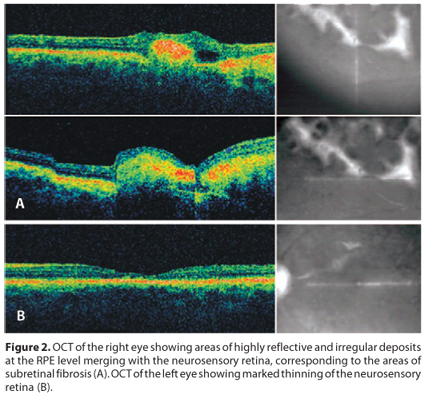
The OCT findings of the right eye showed thinning of the neurosensory retina with irregular thickening of the RPE/choroid layer (Figure 2). There were also multiple areas of highly reflective and irregular nodular deposits at the RPE level merging with the neurosensory retina, corresponding to the areas of subretinal fibrosis. OCT of the left eye showed marked thinning of the neurosensory retina with mild increased choroidal reflectivity. The foveal center of the OS measured 130 µm.
DISCUSSION
The patient described presented with characteristic clinical features of Stargardt's disease including atrophic maculopathy and flavimaculatus yellow flecks. Atypical fundus appearances have been described and include cicatricial chorioretinitis, subretinal fibrosis, subretinal neovascularization and retinal pigment epithelial hyperplasia(1,8,9).
Del Buey et al.(9) described a case of a 14-year-old with diffuse subretinal fibrosis and marked atrophy of the RPE in one eye with Stargardt's disease after a history of mild ocular trauma with a bottle's cork. They suggested that the damaged RPE cells could explain the fibroglial reaction of the retina and pigment migration that occurred as a non-specific response to an inflammatory condition.
De Laey and Verougstraete(1) reported three patients with the characteristics of Stargardt's disease who during follow-up developed massive subretinal fibrosis in the fundus of at least one eye. In two patients, they found massive amounts of orange pigment, presumed to be lipofuscin, in the posterior pole. One of these patients had a history of ocular contusion one week prior to presentation, but no signs of ocular trauma were noted at the time of the exam. In both patients, the orange pigment gradually resorbed leaving extensive areas of subretinal fibrosis and RPE hyperplasia . A third patient who presented with subretinal fibrosis at the initial exam was also described, although it was not definite if the fibrosis was preceded by clinical appearance of orange pigment. The authors speculated that an external factor such as a minor ocular trauma would be capable to provoke a dramatic response in an already diseased RPE. However, there were only limited evidences to confirm that ocular trauma originated their cases.
In 1991, Ko et al.(10) reported histologic evidence that trauma may play a role in releasing massive amounts of lipofuscin from the RPE. He observed a massive accumulation of lipofuscin, using light and electron microscopy, in the retinal pigment epithelium at the posterior pole of a human eye that had been severely traumatized 9 months before enucleation. They suggested that the accumulation of lipofuscin might be originated due to an overload of the RPE cells caused by photoreceptor debris at the time of the trauma.
In conclusion, our case had ophthalmoscopic and angiographic findings which were classic for Stargardt's disease on both eyes and massive amount of subretinal fibrosis on the right eye which was seen at the initial exam. Despite the negative family history and the fact that the patient presented 4 months after the blunt ocular trauma, we believe that it might have played a role as the possible cause of disrupting the diseased RPE with excessive amounts of lipofuscin, leading to the formation of subretinal fibrosis and RPE hyperplasia.
Therefore, we suggest that patients with the diagnosis of Stargardt's disease should be advised on the possible risks of even mild ocular trauma, as well as the possible consequences, which can be prevented by the use of appropriate eye protection during sports and recreational activities as endorsed by the American Academy of Pediatrics and American Academy of Ophthalmology.
REFERENCES
1. De Laey JJ, Verougstraete C. Hiperlipofuscinosis and subretinal fibrosis in Stargardt's disease. Retina. 1995;15(5):399-406.
2. Zhang K, Garibaldi DC, Kniazeva M, Albini T, Chiang MF, Kerrigan M, et al. A novel mutation in the ABCR gene in four patients with autosomal recessive Stargardt disease. Am J Ophthalmol. 1999;128(6):720-4.
3. Briggs CE, Rucinski D, Rosenfeld PJ, Hirose T, Berson EL, Dryja TP. Mutations in ABCR (ABCR4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci. 2001;42(10):2229-36.
4. Simonelli F, Testa F, de Crecchio G, Rinaldi E, Hutchinson A, Atkinson A, et al. New ABCR mutations and clinical phenotype in Italian patients with Stargardt disease. Invest Ophthalmol Vis Sci. 2000;41(3):892-7.
5. Weber BH, Sander S, Kopp C, Walker D, Eckstein A, Wissinger B, et al. Analysis of 21 Stargardt's disease families confirms a major locus on chromosome 1p with evidence for non-allelic heterogeneity in a minority of cases. Br J Ophthalmol. 1996;80(8):745-9.
6. Ober RR, Limstrom SA, Simon RM. Traumatic retinopathy in Stargardt's disease. Retina. 1997;17(3):251-4.
7. Delori FC, Staurenghi G, Arend O, Dorey CK, Goger DG, Weiter JJ. In vivo measurement of lipofuscin in Stargardt's disease - Fundus flavimaculatus. Invest Ophthalmol Vis Sci. 1995; 36(11):2327-31.
8. Parodi MB. Progressive subretinal fibrosis in fundus flavimaculatus. Acta Ophthalmol (Copenh). 1994;72(2):260-4.
9. Del Buey MA, Huerva V, Minguez E, Cristobal JA, Iturbe F, Palomar A. Posttraumatic reaction in a case of fundus flavimaculatus with atrophic macular degeneration. Ann Ophthalmol. 1993;25(6):219-21.
10. Ko MK, Lee WR, McKechnie NM, Hall-Parker B. Post-traumatic hyperlipofuscinosis in the human retinal pigment epithelium. Br J Ophthalmol. 1991;75(1):54-60.
 Corresponde address:
Corresponde address:
Alexandre Achille Grandinetti
Av. Vicente Machado, 1171
Curitiba (PR) - 80420-011 - Brazil
E-mail: [email protected]
Submitted for publication: April 23, 2010
Accepted for publication: August 9, 2011
Study carried out at the Departamento de Oftalmologia e Otorrinolaringologia do Hospital das Clínicas da Universidade Federal do Paraná - UFPR - Curitiba (PR) - Brazil.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: A.A.Grandinetti, None; E.Portella, None; J.Arana, None; N.T.V.Iskorostenski, None.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.