Arq. Bras. Oftalmol. 2011; 74 (6): 10.1590/S0004-27492011000600004
Total: 1409
Jacqueline Martins de Sousa1; Virgínia Fernandes Moça Trevisani2; Rodrigo Pilon Modolo3; Luís Alexandre Rassi Gabriel3; Luis Antonio Vieira4; Denise de Freitas4
DOI: 10.1590/S0004-27492011000600004
ABSTRACT
INTRODUCTION: Scleritis is a rare, progressive and serious disease, the signs of which are inflammation and edema of episcleral and scleral tissues and is greatly associated with systemic rheumatoid diseases. PURPOSE: To perform a prospective and comparative study between ophthalmologic manifestations, serologic findings and therapeutic response of patients with isolated scleritis and scleritis associated with systemic rheumatoid disease. METHODS: Thirty-two outpatients with non-infectious scleritis were studied, from March 2006 to March 2008. The treatment was corticoid eye drops associated with anti-inflammatory agents, followed by systemic corticoids and immunosuppressive drugs if necessary, was considered successful after six months without scleritis recurrence. RESULTS: Fourteen of 32 patients had scleritis associated with systemic rheumatoid disease, of which nine had rheumatoid arthritis, two systemic lupus erythematosus, one Crohn's disease, one Behçet's disease and one gout. There were no difference in relation to involvement and ocular complications, there was predominance of nodular anterior scleritis and scleral thinning was the most frequent complication. The scleritis associated with systemic rheumatoid disease group had 64.3% of autoantibodies, versus 27.8% among those with isolated scleritis and this difference was statistically significant. In the isolated scleritis group 16.7% used anti-inflammatory, 33.3% corticosteroids, 27.8% corticosteroids with one immunosuppressive drug, 5.5% two immunosuppressive drugs, 16.7% corticosteroids with two immunosuppressive drugs and 33.3% pulse of immunosuppressive drugs, there was remission in 88.9%. In the scleritis associated with systemic rheumatoid disease group 7.1% used anti-inflammatory, 7.1% corticosteroids, 50% corticosteroids with one immunosuppressive drug, 7.1% two immunosuppressive drugs and 22.2% pulse of immunosuppressive drugs, 100% had treatment success. CONCLUSION: Prevalence of unilateral nodular scleritis was noted in both groups and higher rates of all the parameters tested were noted in the scleritis associated with systemic rheumatoid disease group. There were no differences between the groups with respect to the use of immunosuppressive drugs and therapeutic response, which was fully satisfactory in the scleritis associated with systemic rheumatoid disease group and satisfactory in the isolated scleritis group.
Keywords: Scleritis; Rheumatic diseases; Autoantibodies; Inflammation; Immunosuppressive agents
RESUMO
INTRODUÇÃO: Esclerite é uma doença grave, rara e progressiva, que envolve inflamação e edema dos tecidos episcleral superficial, profundo e escleral e está associada com doenças sistêmicas reumatológicas em muitos casos. OBJETIVOS: Realizar um estudo prospectivo comparativo entre as manifestações oftalmológicas, achados sorológicos e resposta terapêutica de pacientes com esclerite isolada e com esclerite associada a doenças sistêmicas reumatológicas. MÉTODOS: Trinta e dois pacientes com esclerite não infecciosa participaram do estudo, de março de 2006 a março de 2008. O tratamento realizado baseou-se no uso de colírios de corticoides associados aos anti-inflamatórios não-hormonais, seguidos de corticoides sistêmicos e imunossupressores, se necessário. O sucesso do tratamento foi considerado como seis meses sem crises de esclerite. RESULTADOS: Quatorze dos 32 pacientes apresentaram esclerite associada à doença sistêmica, dos quais nove com artrite reumatóide, dois com lúpus eritematoso sistêmico, um com doença de Crohn, um com doença de Behçet e um com gota. Não houve diferenças em relação ao envolvimento ocular e suas complicações, predominando a esclerite anterior nodular e o afinamento escleral, respectivamente. O grupo com esclerite associada a doenças sistêmicas apresentou 64,3% de positividade de autoanticorpos contra 27,8% no grupo com esclerite isolada, sendo tal diferença estatisticamente significante. No grupo com esclerite isolada, 16,7% fez uso de apenas anti-inflamatórios, 33,3% de corticoide sistêmico, 27,8% de corticoide com um imunossupressor, 5,5% dois imunossupressores, 16,7% corticoide com dois imunossupressores e 33,3% pulsoterapia com imunossupressor; sendo que houve sucesso do tratamento em 88,9%. No grupo com esclerite associada à doença sistêmica, 7,1% fez uso de anti-inflamatórios, 7,1% corticoide sistêmico, 50% corticoide com um imunossupressor, 7,1% dois imunossupressores e 22,2% pulsoterapia com imunossupressor; com 100% de sucesso no tratamento nesse grupo. CONCLUSÃO: Em ambos os grupos houve predomínio da esclerite nodular unilateral e o grupo com esclerite associada a doença sistêmica apresentou taxas maiores de todos os autoanticorpos testados. Não houve diferença entre os grupos em relação ao uso de imunossupressores e à resposta terapêutica, a qual foi totalmente satisfatória no grupo com esclerite associada à doença sistêmica e satisfatória no grupo com esclerite isolada.
Descritores: Esclerite; Doenças reumáticas; Autoanticorpos; Inflamação; Imunossupressores
ORIGINAL ARTICLE ARTIGO ORIGINAL
Comparative study of ophthalmological and serological manifestations and the therapeutic response of patients with isolated scleritis and scleritis associated with systemic diseases
Estudo comparativo entre as manifestações oftalmológicas, sorológicas e resposta terapêutica de pacientes com esclerite isolada e esclerite associada a doenças sistêmicas
Jacqueline Martins de SousaI; Virgínia Fernandes Moça TrevisaniII; Rodrigo Pilon ModoloIII; Luís Alexandre Rassi GabrielIII; Luis Antonio VieiraIV; Denise de FreitasIV
IMedical student, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil
IIProfessor, Universidade de Santo Amaro - UNISA - São Paulo (SP), Brazil; Disciplina de Medicina de Urgência e Medicina Baseada em Evidências, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil
IIIPhysician, Setor de Doenças Externas Oculares e Córnea do Departamento de Oftalmologia da Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil
IVPhysician, Setor de Doenças Externas Oculares e Córnea, Departamento de Oftalmologia, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil
ABSTRACT
INTRODUCTION: Scleritis is a rare, progressive and serious disease, the signs of which are inflammation and edema of episcleral and scleral tissues and is greatly associated with systemic rheumatoid diseases.
PURPOSE: To perform a prospective and comparative study between ophthalmologic manifestations, serologic findings and therapeutic response of patients with isolated scleritis and scleritis associated with systemic rheumatoid disease.
METHODS: Thirty-two outpatients with non-infectious scleritis were studied, from March 2006 to March 2008. The treatment was corticoid eye drops associated with anti-inflammatory agents, followed by systemic corticoids and immunosuppressive drugs if necessary, was considered successful after six months without scleritis recurrence.
RESULTS: Fourteen of 32 patients had scleritis associated with systemic rheumatoid disease, of which nine had rheumatoid arthritis, two systemic lupus erythematosus, one Crohn's disease, one Behçet's disease and one gout. There were no difference in relation to involvement and ocular complications, there was predominance of nodular anterior scleritis and scleral thinning was the most frequent complication. The scleritis associated with systemic rheumatoid disease group had 64.3% of autoantibodies, versus 27.8% among those with isolated scleritis and this difference was statistically significant. In the isolated scleritis group 16.7% used anti-inflammatory, 33.3% corticosteroids, 27.8% corticosteroids with one immunosuppressive drug, 5.5% two immunosuppressive drugs, 16.7% corticosteroids with two immunosuppressive drugs and 33.3% pulse of immunosuppressive drugs, there was remission in 88.9%. In the scleritis associated with systemic rheumatoid disease group 7.1% used anti-inflammatory, 7.1% corticosteroids, 50% corticosteroids with one immunosuppressive drug, 7.1% two immunosuppressive drugs and 22.2% pulse of immunosuppressive drugs, 100% had treatment success.
CONCLUSION: Prevalence of unilateral nodular scleritis was noted in both groups and higher rates of all the parameters tested were noted in the scleritis associated with systemic rheumatoid disease group. There were no differences between the groups with respect to the use of immunosuppressive drugs and therapeutic response, which was fully satisfactory in the scleritis associated with systemic rheumatoid disease group and satisfactory in the isolated scleritis group.
Keywords: Scleritis; Rheumatic diseases; Autoantibodies; Inflammation; Immunosuppressive agents
RESUMO
INTRODUÇÃO: Esclerite é uma doença grave, rara e progressiva, que envolve inflamação e edema dos tecidos episcleral superficial, profundo e escleral e está associada com doenças sistêmicas reumatológicas em muitos casos.
OBJETIVOS: Realizar um estudo prospectivo comparativo entre as manifestações oftalmológicas, achados sorológicos e resposta terapêutica de pacientes com esclerite isolada e com esclerite associada a doenças sistêmicas reumatológicas.
MÉTODOS: Trinta e dois pacientes com esclerite não infecciosa participaram do estudo, de março de 2006 a março de 2008. O tratamento realizado baseou-se no uso de colírios de corticoides associados aos anti-inflamatórios não-hormonais, seguidos de corticoides sistêmicos e imunossupressores, se necessário. O sucesso do tratamento foi considerado como seis meses sem crises de esclerite.
RESULTADOS: Quatorze dos 32 pacientes apresentaram esclerite associada à doença sistêmica, dos quais nove com artrite reumatóide, dois com lúpus eritematoso sistêmico, um com doença de Crohn, um com doença de Behçet e um com gota. Não houve diferenças em relação ao envolvimento ocular e suas complicações, predominando a esclerite anterior nodular e o afinamento escleral, respectivamente. O grupo com esclerite associada a doenças sistêmicas apresentou 64,3% de positividade de autoanticorpos contra 27,8% no grupo com esclerite isolada, sendo tal diferença estatisticamente significante. No grupo com esclerite isolada, 16,7% fez uso de apenas anti-inflamatórios, 33,3% de corticoide sistêmico, 27,8% de corticoide com um imunossupressor, 5,5% dois imunossupressores, 16,7% corticoide com dois imunossupressores e 33,3% pulsoterapia com imunossupressor; sendo que houve sucesso do tratamento em 88,9%. No grupo com esclerite associada à doença sistêmica, 7,1% fez uso de anti-inflamatórios, 7,1% corticoide sistêmico, 50% corticoide com um imunossupressor, 7,1% dois imunossupressores e 22,2% pulsoterapia com imunossupressor; com 100% de sucesso no tratamento nesse grupo.
CONCLUSÃO: Em ambos os grupos houve predomínio da esclerite nodular unilateral e o grupo com esclerite associada a doença sistêmica apresentou taxas maiores de todos os autoanticorpos testados. Não houve diferença entre os grupos em relação ao uso de imunossupressores e à resposta terapêutica, a qual foi totalmente satisfatória no grupo com esclerite associada à doença sistêmica e satisfatória no grupo com esclerite isolada.
Descritores: Esclerite; Doenças reumáticas; Autoanticorpos; Inflamação; Imunossupressores
INTRODUCTION
Scleritis is a rare, progressive and serious disease, the signs of which are inflammation and edema of superficial episcleral tissues. Clinical and epidemiological characteristics include: severe ocular pain radiating to the ipsilateral side, redness of the sclera and conjunctiva, sometimes changing to a purple hue, affects mostly young and middle aged women and is generally bilateral(1). This disease is difficult to treat and can progress with serious complications if not done adequately(2,3). Identifiable causes for scleritis include surgical trauma, bacterial, viral and parasitic infections, autoimmune diseases and primary vasculitis. However, an underlying systemic disease is not identified for the vast majority of cases of scleritis, even after clinical and laboratory tests(4). Scleritis is greatly associated with systemic rheumatoid disease (30 to 50%), especially rheumatoid arthritis (RA), Wegener's granulomatosis, nodular polyarthritis and systemic lupus erythematosus (SLE)(5-7). Rarely scleritis is a part of the systemic involvement of infectious diseases (about 5 to 10%)(8).
Patients with scleritis do not usually respond well to topical therapies involving corticoids and non hormonal anti-inflammatory drugs (NSAIDs). However, some patients respond well to the non hormonal anti-inflammatory drugs, and these medications are prescribed for the initial treatment of non necrotizing scleritis. A great number of cases of scleritis require the use of systemic corticoids and about 25% require the use of associated immunosuppressive agents to control the inflammation(2,9-11).
The aim of this work was to perform a comparative study between ophthalmologic manifestations, serologic findings and therapeutic response of patients with isolated scleritis and scleritis associated with systemic rheumatoid disease.
METHODS
CASUISTIC
Thirty-two outpatients were consecutively selected from the External Disease and Cornea sector of the Department of Ophthalmology, UNIFESP. Patients from both genders diagnosed with non-infectious scleritis were included from the period of March 2006 to March 2008. Patients who gave consent for participation were submitted to a protocol consisting of clinical and ophthalmologic evaluation. We used the criteria established by Watson et al.(12) for scleritis diagnosis and classification: anterior diffuse scleritis, nodular scleritis, necrotizing scleritis and posterior scleritis. In order to diagnose rheumatologic disease, all patients were evaluated by the same rheumatologist, and systemic disease diagnosis was carried out according to The American College of Rheumatology Classification Criteria specific for each disease(13). The time period of the disease was defined as the period from the first scleritis-related clinical symptom to the present date.
METHODOLOGY
Laboratory examinations were performed to test for autoantibodies as follows: antinuclear antibodies (ANA) were assayed using the indirect immunofluorescence assay (IFA) with HEp-2 cells as the substrate (Hemagen, Whalton, MD)(14), rheumatoid factor (RF) was assayed using the latex particle agglutination test(15), anti-neutrophil cytoplasmic antibody, c-ANCA and p-ANCA were assayed using the human neutrophil IIF technique(16), and anti-perinuclear factor (APF) antibodies were assayed using the protocol proposed by Hoet(17) for IIF. Other examinations performed were: erythrocyte sedimentation rate using the Westergren technique, complete blood count and urine type I test. Some patients were submitted to chest and hand X-rays when necessary.
TREATMENT
The scleritis treatment organogram from the Cornea and External Disease ambulatory (Department of Ophthalmology, UNIFESP) was followed. This treatment starts with corticoid eye drops associated with NSAID, followed by the use of systemic corticoids(18). Patients who continued to have eye inflammation (red eye or eye pain) were further treated with immunosuppressors such as methotrexate (MTX) 7.5 to 15 mg/week, azathioprine (AZA) 100 mg/day, cyclosporine A (CsA) 3.0 to 5.0 mg/kg/day, mycophenolate mofetil 2 g/day, clorambucil 0.1 mg/kg/day, leflunomide 20 mg/day or cyclophosphamide pulse therapy of 15/mg/day once a month for four months(19). Due to the varying degree of disease severity, the organogram was not always followed. The organogram was also not followed in the case of patients with other associated rheumatic diseases already undergoing treatment with corticoids and/or immunosuppressors. In a few more severe cases, the patients received pulse therapy with corticoids, subconjunctival corticoids or biological therapy (adalimumab)(20-22).
The treatment was considered successful after a period of six months without scleritis recurrences and without clinical symptoms.
The data collected from the clinical and laboratory tests were evaluated for statistical analysis. The following statistical analysis were carried out: Mann-Whitney test for comparing groups according to age, chi-squared test and the Fisher exact test for analyzing the data presented in 2x2 tables. Chi-square tests were performed using a rejection level of p<0.05 (or 5%), and significant values are marked with an asterisk.
RESULTS
A total of 32 patients diagnosed with scleritis were evaluated at the External Diseases and Cornea ambulatory from the Department of Ophthalmology, UNIFESP during the period of this study. Clinical evaluation and laboratory tests diagnosed 18 (56.25%) patients with isolated scleritis (IS) and 14 (43.75%) patients with scleritis associated with systemic disease (SASD), of which 9 had RA, 2 had SLE, 1 had Crohn's disease, 1 had Behçet's disease and 1 had gout.
The average duration of the SASD group was 6.8 years, ranging from zero to 33 years. The average age for the IS group was 58.4 years old (ranging from 25 to 79 years), and was slightly higher than the average age for the SASD group which was 54.2 years old (ranging from 16 to 81 years). The female gender predominated for both groups; 66.7% and 78.6%, respectively.
Characteristics of the afflicted eye are displayed in table 1. No differences can be observed between groups regarding eye involvement patterns, and in both cases, there's a predominance of anterior nodular scleritis.
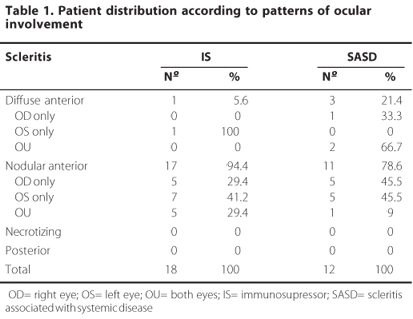
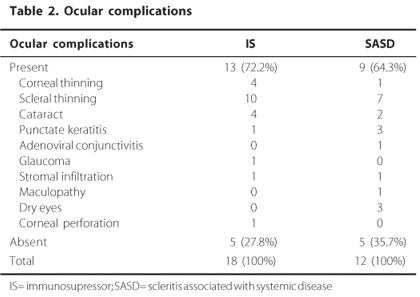
The average duration of the eye disease was 4.9 years for the IS group (ranging from 2 to16 years) and 3 for the SASD group (ranging from 1 to 6 years). Eye complications presented by the patients during the course of the disease are summarized in table 2. No differences were found between groups regarding the presence of complications or considering the type of complications presented, and scleral thinning was the most frequent eye alteration found for both groups.
Laboratory tests showed that autoantibodies were present although no differences were observed between groups. However, autoantibody titres varied between groups. Three patients in the IS group were ANA positive (all with titres of 1/80 - fine speckled pattern), one was RF positive (1/80) and one was p-ANCA positive (1/80). In the SASD group, four patients were positive for ANA (titres of 1/1280 - coarse speckled pattern, 1/1280 - homogeneous pattern, 1/640 - fine speckled pattern and 1/80 - coarse standard dotted), four cases were RF positive (1/640, 1-64, 86.10 and 74), two patients were p-ANCA positive and two were APF positive.
TREATMENT USED FOR THE PATIENTS WITH ISOLATED SCLERITIS
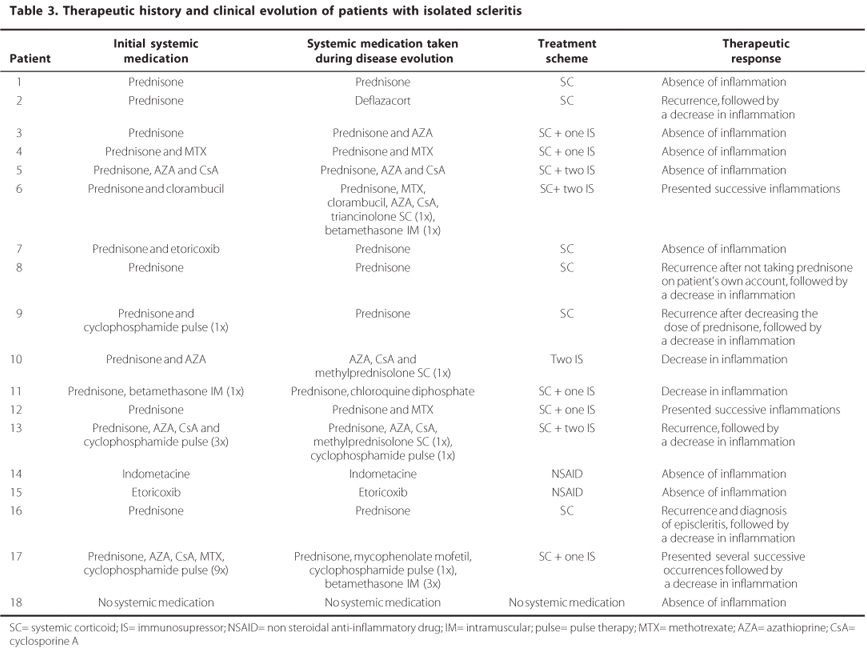
Systemic medication was used for the treatment of scleritis in 17 of the 18 patients. A summary of the treatments used in this group are listed in table 3, which also shows the medications used before and after the patients enrolled in the External Diseases and Cornea ambulatory, from the Department of Ophthalmology, UNIFESP.
The success rate for this treated group was 88.9%, therefore in two patients recurrence of the scleritis occurred in less than six months. Recurrence occurred in the first patient once the dose of corticoids was reduced to less than 20 mg/day, even when associated with other immunosuppressors such as clorambucil, AZA and MTX, and occurred in the second patient after taking lower doses of corticoids, even when associated with MTX.
TREATMENT USED FOR PATIENTS WITH SCLERITIS ASSOCIATED WITH SYSTEMIC DISEASE
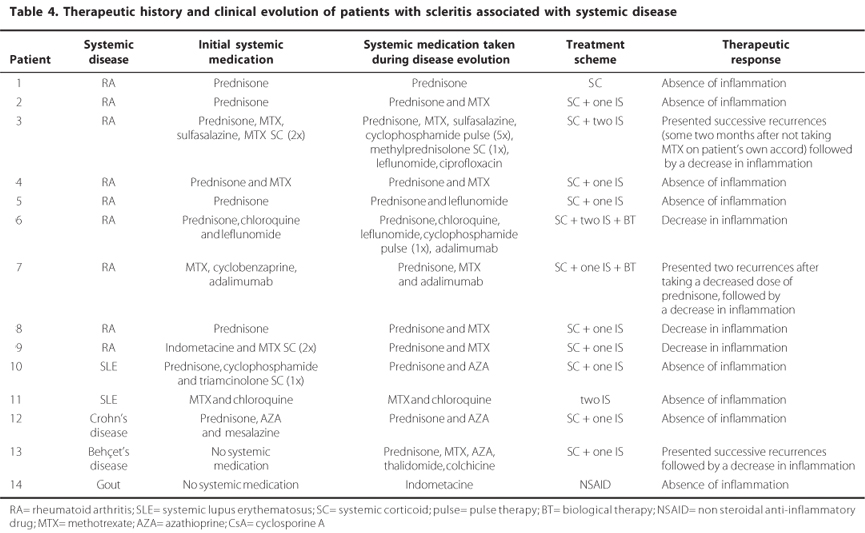
Only three of the fourteen patients with SASD were not already taking systemic medication when diagnosed with scleritis. After diagnostic testing, RA, Behçet's disease and gout were confirmed in these patients, while the other patients had already been diagnosed and were undergoing treatment for their respective scleritis associated systemic diseases. Even though the patients were taking corticoids and/or immunosuppressors, they all presented inflammation of the sclera. Table 4 summarizes the distribution of the systemic diseases, the treatments used and the medication the patients were taking before and after being enrolled in the ambulatory practices of the External Diseases and Cornea Sector, from the Department of Ophthalmology, UNIFESP.
One of the patients with RA presented secondary systemic infection from the use of immunosuppressors and was treated with 500 mg ciprofloxacin twice a day for seven days.
An alternative scheme was used for the patient with Behçet's disease; AZA associated with prednisone, which controlled the patient's clinical ocular manifestations. Later on, both medications were discontinued due to gastric intolerance, and substituted with thalidomide, which controlled mucous manifestations (oral and genital). This patient developed neuritis as a result of the medication, which was then suspended. Subsequently, this patient suffered an ocular crisis, muscular pains and reappearance of oral and genital ulcers which were treated with prednisone and AZA.
In this group, recurrence did not occur in any of the patients during the six months following the end of the treatment, which therefore characterises this treatment as 100% successful.
ADVERSE EFFECTS
One patient from the IS group presented side effects due to the use of prednisone, one due to AZA and one due to the use of CsA and AZA. One patient from the SASD group presented side effects due to the use of AZA.
DISCUSSION
Fourteen of the thirty-two patients studied (43.75%) had an underlying scleritis associated systemic disease, a higher percentage than that found in the recent literature(23,24).
The female gender prevailed in both groups involved in this study; 66.7% in the IS group and 78.6% in the SASD group. Literature also shows a prevalence of female patients(3,25).
The average age of patients in the IS group was higher than that of the SASD group; 58.4 and 54.2 years old, respectively. These averages were higher than and diverged from those found in recent literature, which shows a higher average age for patients in the SASD group compared to the IS group; 55.24 and 48.29 years old, respectively(6).
In regards to ocular manifestations, unilateral nodular scleritis predominated in both groups. Recent literature shows a prevalence of diffuse scleritis, however with unilateral involvement prevailing(6). After extensive research in scientific literature, we could not find a scale or classification for ocular inflammation, thus, we used the clinical symptoms (red eye and ocular pain) as parameters for improvement or worsening of the ocular inflammation. In the same way, we could not find a consensus in the literature about the period in which the patient must be asymptomatic for considering treatment success, then, we considered six months, by our clinical experience.
Patients in the IS group had been diagnosed with ocular disease for a longer period of time than those from the SASD group; 4.9 and 3 years, respectively.
In the IS group, 72.2% presented ocular complications and in the SASD group, 64.3%; however this difference did not show statistical significance. Scleral thinning prevailed in both groups, which is in accordance with previous studies in literature(3). In our study, all the cases of cataract were caused by de use of corticoids and the cases of corneal perforation were related to corneal melting.
The prevailing systemic disease in the SASD group was AR (64.4%). This finding is in accordance with literature which identifies RA as the main systemic disease associated with scleritis(1).
There were statistically significant differences between the two groups regarding the laboratory tests; the SASD group indicated a higher proportion of positive results for the parameters tested, this is an important finding that should be considered in the investigation of patients with scleritis. This group presented positive ANA in 28.6% of the cases, positive RF in 28.6%, positive APF in 14.3% and p-ANCA positive in 14.3%. The IS group presented positive ANA in 16.7% of the cases, positive RF in 5.5% and positive p-ANCA in 5.5%. Data from the literature show that a significant percentage of patients with scleritis are positive for ANCA, and these patients are more likely to have severe ocular disease and undiagnosed systemic disease associated(26,27). APF is considered an important marker for the diagnosis of rheumatoid arthritis(28). The initial treatment for scleritis is based on controlling the ocular inflammatory crisis. As such, protocol establishes an initial treatment with NSAID, followed by systemic corticoids and immunosuppressive agents in more complicated cases in order to control the disease(29).
Regarding the behavior of scleritis and therapeutic response, both groups showed similar results, independently of the systemic disease. Patients with and without associated systemic disease were likely to require systemic therapy (94.4% and 100%, respectively), as related in recent literature(30). During the therapeutic follow-up of IS patients, the medications used during a crisis were maintained, trying to reduce or even suspend at first the use of systemic corticoids, followed by the immunosuppressors(8,31-34). For the treatment of SASD the aim is to change the initial treatment according to the underlying disease, and in some cases adequate treatment for the systemic disease already improves the ocular disease(4,35). Alternatively, biological therapies may be used for cases that do not respond to these treatment schemes(36,37).
CONCLUSION
In regards to ocular manifestations, we observed a prevalence of unilateral nodular scleritis in both groups. Serologically, we observed higher positive results for all laboratory parameters tested in the SASD group. The therapeutic contribution was greatly satisfactory for the SASD group with 100% therapeutic success, while the IS group presented an 88.9%. In this study, there were no differences between the two groups in relation to the eye compromising and the therapeutic response.
REFERENCES
1. Watson PG, Hazlemaan BL, Pavesio CE. The sclera and systemic disorders. 2nd ed. London: Butterworth-Heinemann; 2004.
2. Smith JR, Mackensen F, Rosenbaum JT. Therapy insight: scleritis and its relationship to systemic autoimmune disease. Nat Clin Pract Rheumatol. 2007;3(4):219-26.
3. Tuft SJ, Watson PG. Progression of scleral disease. Ophthalmology. 1991;98(4):467-71.
4. Pavesio CE, Meier FM. Systemic disorders associated with episcleritis and scleritis. Curr Opin Ophthalmol. 2001;12(6):471-8. Review.
5. Watson PG, Young RD. Scleral structure, organisation and disease. A review. Exp Eye Res. 2004;78(3):609-23.
6. Sainz de la Maza M, Foster CS, Jabbur NS. Scleritis associated with systemic vasculitic diseases. Ophthalmology. 1995;102(4):687-92.
7. Hamideh F, Prete PE. Ophthalmologic manifestations of rheumatic diseases. Semin Arthritis Rheum. 2001;30(4):217-41. Review.
8. Albini TA, Rao NA, Smith RE. The diagnosis and management of anterior scleritis. Int Ophthalmol Clin. 2005;45(2):191-204.
9. Sainz de la Maza M, Jabbur NS, Foster CS. An analysis of therapeutic decision for scleritis. Ophthalmology. 1993;100(9):1372-6.
10. Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol. 2000;130(4):469-76.
11. Carrasco MA, Cohen EJ, Rapuano CJ, Laibson PR. Therapeutic decision in anterior scleritis: our experience at a tertiary care eye center. J Fr Ophtalmol. 2005;28(10):1065-9.
12. Watson PG, Hayreh SS. Scleritis and episcleritis. Br J Ophthalmol. 1976;60(3):163-91.
13. Thorne JE, Jabs DA. The rheumatic diseases. In: Tasman W, Jaegar EA, editors. Duane's clinical ophthalmology. Vol 5. Philadelphia: Lippincott, Williams & Wilkins; 2009.
14. Friou GJ. Clinical application of a test for lupus globulin-nucleohistone interaction using fluorescent antibody. Yale J Biol. 1958;31(1):40-7.
15. Plotz CM, Singer JM. The latex fixation test. I. Application to the serologic diagnosis of rheumathoid arthritis. Am J Med. 1956;21(6):888-92.
16. Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c-ANCA) testing in the diagnosis of Wegener granulomatosis. A literature review and meta-analysis. Ann Intern Med. 1995;123(12):925-32. Comment in Ann Intern Med. 1996;125(7):622; author reply 622-3. ACP J Club. 1996; 124(2):48.
17. Hoet RMA. Detection of antiperinuclear factor and antikeratin antibodies. In: Van Venrooij WJ, Maini RN, editors. Manual of biological markers of disease. New York: Kluwer Academic Publishers; 1993.
18. Santos NC, Sousa LB, Trevisani VFM, Freitas D, Vieira LA. Manifestações destrutivas da córnea e esclera associadas a doenças do tecido conectivo: relato de 9 casos. Arq Bras Oftalmol. 2004;67(4):675-80.
19. Hemady R, Tauber J, Foster S. Immunosuppressive drugs in immune and inflammatory ocular disease. Surv Ophthalmol. 1991;35(5):369-85. Erratum in Surv Ophthalmol. 1991; 35(6):475. Dosage error in article text.
20. Bertele' V, Assisi A, Di Muzio V, Renzo D, Garattini S. New antirheumatic drugs: any real added value? A critical overview of regulatory criteria for their marketing approval. Eur J Clin Pharmacol. 2007;63(9):879-89.
21. Li EK, Tam LS, Tomlinson B. Leflunomide in the treatment of rheumatoid arthritis. Clin Ther. 2004;26(4):447-59. Review.
22. Alonso-Ruiz A, Pijoan JI, Ansuategui E, Urkaregi A, Calabozo M, Quintana A. Tumor necrosis factor alpha drugs in rheumatoid arthritis: systematic review and metaanalysis of efficacy and safety. BMC Musculoskelet Disord. 2008;9:52.
23. Akpek EK, Thorne JE, Qazi FA, Do DV, Jabs DA. Evaluation of patients with scleritis for systemic disease. Ophthalmology. 2004;111(3):501-6. Comment in Ophthalmology. 2007; 114(6):1232.
24. Erkanli L, Akova YA, Guney-Tefekli E, Tugal-Tutkun I. Clinical features, prognosis, and treatment results of patients with scleritis from 2 tertiary eye care centers in Turkey. Cornea. 2010;29(1):26-33.
25. Lachmann SM, Hazleman BL, Watson PG. Scleritis and associated disease. Br Med J. 1978;1(6105):88-90.
26. Hoang LT, Lim LL, Vaillant B, Choi D, Rosenbaum JT. Antineutrophil cytoplasmic antibody-associated active scleritis. Arch Ophthalmol. 2008;126(5):651-5.
27. Lin P, Bhullar SS, Tessler HH, Goldstein DA. Immunologic markers as potential predictors of systemic autoimmune disease in patients with idiopathic scleritis. Am J Ophthalmol. 2008;145(3):463-71.
28. Aragaki WK, Sousa LB, Trevisani VFM, Fuzzi H, Andrade LEC. Sclera-specific and non-sclera-specific autoantibodies in the serum of patients with non-infectious anterior scleritis. Rev Bras Reumatol. 2007;47(3):173-9.
29. Höfling-Lima AL, Moeller CTA, Freitas D, et al. Esclerites e episclerites. In: Moeller CTA, Freitas D, Höfling-Lima AL, Martins ENM, editores. Manual de condutas em oftalmologia. São Paulo: Atheneu; 2008. p.297-9.
30. Raiji VR, Palestine AG, Parver DL. Scleritis and systemic disease association in a community-based referral practice. Am J Ophthalmol. 2009;148(6):946-50.
31. Pujari SS, Kempen JH, Newcomb CW, Gangaputra S, Daniel E, Suhler EB, et al. Cyclophosphamide for ocular inflammatory diseases. Ophthalmology. 2010;117(2):356-65.
32. Daniel E, Thorne JE, Newcomb CW, Pujari SS, Kaçmaz RO, Levy-Clarke GA, et al. Mycophenolate mofetil for ocular inflammation. Am J Ophthalmol. 2010;149(3):423-32.e1-2.
33. Jachens AW, Chu DS. Retrospective review of methotrexate therapy in the treatment of chronic, noninfectious, nonnecrotizing scleritis. Am J Ophthalmol. 2008;145(3):487-92.
34. Pasadhika S, Kempen JH, Newcomb CW, Liesegang TL, Pujari SS, Rosenbaum JT, et al. Azathioprine for ocular inflammatory diseases. Am J Ophthalmol. 2009;148(4):500-9.e2.
35. Smith JR, Mackensen F, Rosenbaum JT. Therapy insight: scleritis and its relationship to systemic autoimmune disease. Nat Clin Pract Rheumatol. 2007;3(4):219-26.
36. Michalova K, Lim L. Biologic agents in the management of inflammatory eye diseases. Curr Allergy Asthma Rep. 2008;8(4):339-47.
37. Lim L, Suhler EB, Smith JR. Biologic therapies for inflammatory eye disease. Clin Experiment Ophthalmol. 2006;34(4):365-74. Review.
 Correspondence address:
Correspondence address:
Jacqueline Martins de Sousa
Rua Napoleão de Barros, 874 - Apto. 42
São Paulo (SP) - 04024-002 - Brazil
E-mail: [email protected]
Submitted for publication: February 8, 2011
Accepted for publication: November 5, 2011
Study carried out at the Setor de Doenças Externas Oculares e Córnea, Departamento de Oftalmologia, Universidade Federal de São Paulo - UNIFESP - São Paulo (SP), Brazil.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: J.M.de Sousa, None; V.F.M.Trevisani, None; R.P. Modolo, None; L.A.R.Gabriel, None; L.A.Vieira, None; D.de Freitas, None.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}