Arq. Bras. Oftalmol. 2001; 64 (2): 10.1590/S0004-27492001000200016
Total: 2222
Flávio A. Marigo1; Sebastião Cronemberger4; Nassim Calixto4
DOI: 10.1590/S0004-27492001000200016
Neuroproteção: situação atual no glaucoma
Neuroprotection: current status of glaucoma
Flávio A. Marigo1, 3, 4
Sebastião Cronemberger2, 3
Nassim Calixto2, 3
O glaucoma é uma das três principais causas de cegueira no mundo. Até há pouco tempo, os avanços na terapia antiglaucomatosa concentravam-se na expectativa da obtenção de drogas hipotensoras mais eficazes e com menor incidência de efeitos colaterais. Os avanços recentes proporcionados por um melhor conhecimento da fisiopatologia e principalmente da química molecular da doença têm propiciado o surgimento de novas e promissoras perspectivas de tratamento para a moléstia.
Dentro dessa ótica inovadora, duas linhas de investigação se destacam. A primeira baseia-se no reconhecimento de que a terapia molecular para aumentar a resistência neuronal a um estímulo agressor ("neuroproteção") torna-se cada vez mais tangível. A segunda linha investiga a possibilidade de obter-se a regeneração neuronal de forma controlada ("neuroregeneração"). Essa linha vislumbra ainda inúmeros obstáculos a serem superados.
O emprego de neuroprotetores em medicina não é recente. A investigação dessa nova classe de drogas iniciou-se a partir da melhor compreensão dos fenômenos bioquímicos envolvidos nas lesões do sistema nervoso central (SNC). Observou-se que, a despeito da etiologia da lesão primária, dano subsequente afeta neurônios inicialmente não afetados pelo estímulo agressor. Qualquer que seja esse estímulo (trauma- incluindo o barotrauma, edema, inflamação, isquemia, etc.) alterações bioquímicas sensíveis fazem sentir-se no meio ambiente metabólico dos neurônios vizinhos àqueles inicialmente agredidos, promovendo perda neuronal secundária, mesmo após a cessação da agressão primária. O emprego de drogas capazes de regularizar esse micro-ambiente ou de aumentarem a resistência neuronal ao mesmo ("neuroprotetores") vêm sendo estudado no tratamento de moléstias que afetam o SNC, destacando-se a doença de Parkinson(1-10), acidentes vasculares cerebrais(11-19) e doenças degenerativas(20-22), dentre outras.
Paralelamente, vários trabalhos têm sido realizados objetivando a compreensão desses fenômenos e a possibilidade do emprego dessas drogas em doenças oculares como o glaucoma. Uma discussão a esse respeito faz-se, portanto, necessária.
GLAUCOMA: ETIOPATOGÊNESE
Conceito atual de glaucoma
O glaucoma é modernamente definido como uma neuropatia óptica crônica caracterizada por uma perda progressiva das fibras neurais do nervo óptico podendo culminar na perda completa de suas funções(23-26).
Etiopatogênese
A etiopatogênese do glaucoma permanece objeto de discussão(23, 26). Vários fatores parecem contribuir, ainda que em graus variáveis, para o seu desencadeamento. A pressão intra-ocular (Po) é ainda considerada por muitos autores como o principal fator de risco para a doença porque: a) o glaucoma freqüentemente desenvolve-se em olhos com Po situadas acima dos valores de normalidade(23-26); b) a Po é o único fator de risco de fácil mensuração e tratamento e c) a normalização da Po geralmente retarda ou impede a sua evolução. Não obstante, a resistência demonstrada ao desenvolvimento do glaucoma por alguns indivíduos com hipertensão ocular evidente assim como a sua ocorrência em raros indivíduos com Po normal ou pouco acima dos valores normais ("glaucoma de pressão normal") mostram que a Po não é o único fator significativo para a doença. A tolerância a uma dada Po sofre variação individual e outros fatores tais como: a) aumento da resistência vascular periférica ocular; b) redução do fluxo sangüíneo ao nervo óptico; c) a ação de excitotoxinas e a d) depleção de neurotrofinas para as células ganglionares da retina (CGR) podem interferir, aumentando a predisposição do indivíduo ao glaucoma ou mesmo desencadeando-o(23, 26-27). (Quadro 1; Gráfico 1).

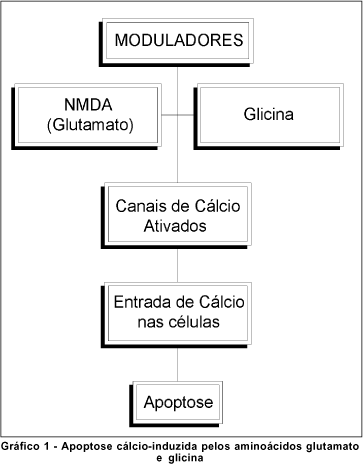
Quaisquer que sejam os fatores envolvidos, a intervenção de um ou mais desses fatores desencadeia e agrava o glaucoma através de duas seqüências de eventos: a lesão neuronal (LN) primária e a LN secundária.
Lesão neuronal primária
A LN primária consiste na perda primária de CGR e de fibras nervosas no nervo óptico (FNNO) causada diretamente pela ação do estímulo agressor (ex.: aumento da Po)(23, 25-26, 28-29). Essa perda ocorre através de três mecanismos: a) mecânico, por compressão neuronal ao nível da lâmina crivosa; b) vascular, por isquemia do nervo óptico decorrente de alterações no fluxo sangüíneo neural e c) axoplasmático, por bloqueio ou retardamento do fluxo axoplasmático ao longo do neurônio (Quadro 2)(23, 25-26, 30).

Lesão neuronal secundária
As CGR e as FNNO acometidas pela LN primária iniciam um processo de degeneração e morte celular com liberação de mediadores químicos para o meio extra-celular que, agindo sobre as células vizinhas inicialmente poupadas pela agressão primária, promovem uma perda adicional de neurônios: é a LN secundária(26). Observa-se pois que: a) mesmo após a interrupção do estímulo primário que desencadeou o glaucoma (por exemplo, pelo controle da Po), dano adicional de neurônios pode ocorrer enquanto esses mediadores persistirem na matriz extra-celular(25, 30-31); b) a extensão da LN secundária é proporcional à extensão da LN primária, uma vez que, quanto mais intensa ou duradoura esta se mostrar, tanto maior será a liberação dos mediadores da LN secundária(23, 25). Esses eventos ocorrem simultaneamente, de tal maneira que, em dado momento, uma mesma região do nervo óptico apresenta células em degeneração pela LN primária, pela LN secundária e ainda células íntegras, poupadas do estímulo agressor.
Apoptose e Excitotoxicidade. Mediadores da lesão neuronal
A perda neuronal, seja causada pela LN primária ou secundária, decorre de dois mecanismos intracelulares: apoptose e excitotoxicidade(27, 30, 32-34). Apoptose consiste na morte celular geneticamente programada, caracterizada por condensação e fragmentação da cromatina nuclear e do citoplasma celular que, envoltos por unidades de membrana celular, são fagocitados por macrófagos ou outras células capazes de fagocitose, sem que haja reação inflamatória(30, 32-33). A apoptose em si é um mecanismo de homeostase normalmente presente em organismos multicelulares, especialmente durante a embriogênese. Todavia, em condições anormais, várias modalidades de estímulos podem ativar a apoptose, tais como: isquemia, irradiação, fatores de crescimento, hormônios e infecções viróticas(30, 32-33).
Excitotoxicidade consiste na liberação maciça de neurotransmissores por células atingidas por um estímulo agressor(31, 34). Essas substâncias atuam como mediadores no desencadeamento de apoptose- são os mediadores da LN. Vários são os mediadores envolvidos, destacando-se, entre eles, o glutamato e a glicina(23-24, 26, 31, 35-41).
O glutamato possui vários receptores de superfície de membrana celular, como o receptor NMDA (N-Metil-D-Aspartato)(26, 34-35, 38). A ligação do glutamato e/ou da glicina aos seus receptores permite a abertura dos canais de cálcio na membrana celular e um influxo desse íon para o interior da célula(26, 34, 38). O cálcio, além de determinar um desequilíbrio iônico e elétrico transmembrana, atua também como um segundo mensageiro, ativando os mecanismos de cascata que culminam em degeneração e morte celular (Gráfico 2)(24,38).
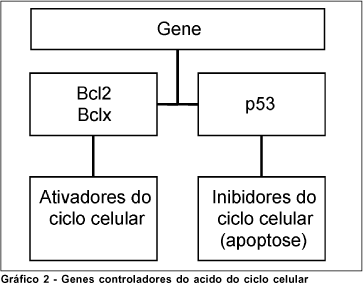
Outras alterações bioquímicas dos meios extra e intra-celular podem atuar como mediadores no desencadeamento dos processos de excitotoxicidade e apoptose, tais como alterações iônicas (elevação do potássio)(26), liberação de radicais livres e de pró-oxidantes que causem "stress" oxidativo (óxido nítrico, superóxido, ferro iônico, derivados da peroxidação lipídica e outros)(26, 42-43, 5, 9, 44-46), de outros neurotransmissores (23-26, 30-31, 34, 37, 40-41), de fatores de crescimento(26, 30) e de mediadores liberados pelas células imunitárias (Quadro 3)(26).

GLAUCOMA: HISTOPATOLOGIA
Em decorrência da perda neuronal por apoptose e excitotoxicidade, decorrentes dos processos de LN primária e secundária, observa-se: a) perda de CGR; b) adelgaçamento da camada de fibras nervosas da retina (CFNR); c) perda de fibras nervosas do nervo óptico (FNNO) e, em casos mais avançados; d) alterações nas células horizontais e perda de fotorreceptores(47-50). É importante rememorar que a CFNR e as FNNO são constituídas pelos axônios neurais das CGR.
NEUROPROTEÇÃO
Neuroproteção consiste em retardar ou impedir a lesão de CGR ainda não afetadas pela LN primária(23, 25-26, 30, 41). Em outras palavras, o tratamento neuroprotetor visa impedir ou retardar a LN secundária sobre as células ainda não afetadas no decurso da doença. Basicamente, a neuroproteção pode ocorrer em três níveis 26:
a) Nível extracelular: pela inativação ou pelo bloqueio dos mediadores da LN secundária e pela modulação das células imunitárias;
b) Superfície de membrana celular: pelo bloqueio dos receptores de superfície das CGR para os mediadores da LN secundária;
c) Meio intracelular: pela ativação dos genes envolvidos na regulação do ciclo celular (compostos antiapoptose).
O conhecimento atual sobre drogas potencialmente neuroprotetoras deriva de estudos sobre neuroproteção a nível de células do SNC. A nível extracelular, o neuroprotetor MK801 (maleato de dizolcipina), o canabinóide HU201 e os glicocorticóides têm uma ação inibidora da morte de CGR induzidas por isquemia(17- 18) bem como alguma capacidade de modulação das células imunitárias(26, 51). Os glicocorticóides parecem exercer também parte de sua ação neuroprotetora pela inibição da peroxidação lipídica(26).
Dentre os inibidores dos mediadores da LN secundária, os inibidores da liberação do glutamato (riluzol), os antagonistas poliamínicos (eliprodil), agentes antioxidantes (selegina(5), dopamina(5), manganês(9)) e os inibidores do óxido nítrico (gangliosídeos e inibidores de radicais livres) têm demonstrado atividade neuroprotetora(26).
A nível de membrana celular, as pesquisas têm se concentrado na tentativa de neuroregulação da apoptose cálcio induzida (Gráfico 1)(26, 34, 52). Este efeito pode ser obtido pelo emprego de agentes bloqueadores do receptor glutamato NMDA (como o MK801, memantina e outros congêneres de geração mais recente) ou pelo uso de antagonistas competitivos do mesmo receptor como o selfotel (CGS19755) e outros antagonistas poliamínicos (CPS101,606)(19, 26). Outras duas possibilidades seriam através da inibição do receptor modulador (Magnésio, memantina e óxido nitroso) ou pela inibição do receptor glicina (kainato)(26).
O emprego de bloqueadores dos canais de cálcio tem sido extensivamente investigado(34). Essas drogas bloqueiam o influxo de íons cálcio no interior da célula, permitindo o re-equilíbrio iônico e de potenciais elétricos transmembranas. Destacam-se a nitradipina, o diltiazem, o verapamil e a nicardipina(26, 52). Entretanto, a possibilidade de efeito dele-tério sobre o fluxo sangüíneo para o nervo óptico, através da redução da pressão de perfusão para o mesmo, faz com que o valor desses medicamentos como agentes neuroprotetores esteja ainda por ser estabelecido.
A nível intra-celular, tem-se investigado o controle do ciclo genético que regula o processo de apoptose. Os grupamentos genéticos Bcl2 e Bclx ativam o ciclo celular(10, 26, 53), ao passo que o grupamento p53 o deprime(26). Portanto, drogas que promovam a ativação dos genes Bcl e a inibição do gene p53 têm ação anti-apoptótica(10, 53). Uma classe de compostos chamados de lazaróides além de determinadas neurotrofinas parece exercer essa ação(26). Uma outra opção, baseada em observações obtidas em condições experimentais, consiste no uso dos agentes neurotróficos. Dentre estes, o bFGF ("Basic Fibroblast Growth Factor") e BDNF ("Brain Derived Neurotrophic Factor") e CNTF ("Ciliary Neurotrophic Factor") mostraram ação anti-apoptótica(54). Entretanto, nenhum efeito foi observado com os agentes NGF ("Nerve Growth Factor"), EGF ("Epidermal Growth Factor") e PDNF ("Platelet Derived Neurothrophic Factor")(26). Uma terceira via de modulação genética consiste no uso de vetores virais para a inoculação de moléculas ativadoras do ciclo celular(1, 55-57). Esses vetores virais, apropriadamente modificados, carreiam "moléculas terapêuticas" (por exemplo seqüências genéticas ativadoras do ciclo celular), para células-alvo no SNC, inoculando no seu interior o material desejado. Essas células, uma vez modificadas geneticamente, passam a ter então capacidade de sintetizar compostos intracelulares capazes de ativar o ciclo celular e impedir a apoptose.
PERSPECTIVAS
Os recentes conhecimentos adquiridos a respeito da etiopatogênese do glaucoma e de várias doenças do SNC, principalmente em seus aspectos bioquímicos, abriram novas perspectivas para a sua abordagem terapêutica. Até recentemente, o tratamento do glaucoma dependia exclusivamente do controle farmacológico ou cirúrgico da Po, seu principal fator de risco. A redução da Po a níveis compatíveis com a preservação do nervo óptico permanece como pilar essencial no tratamento do glaucoma, desde que a Po elevada constitui-se no principal elemento detonador das reações em cascata que desencadeiam a LN primária para posteriormente, prosseguir a cascata da LN secundária, objetivo da intervenção do processo neuroprotetor. A neuroproteção poderá constituir-se na segunda baliza do tratamento contra essa doença, ao impedir a progressão do dano celular aos neurônios não afetados inicialmente pela doença e, portanto, por possibilitar uma melhor manutenção do patrimônio neuronal do indivíduo. Uma terceira via poderá ser constituída pelo estímulo à regeneração neuronal controlada (neuroregeneração) ou pelo transplante de células neuronais.
Não obstante, antes que os neuroprotetores venham a ser utilizados no tratamento do glaucoma, algumas dificuldades deverão ser superadas. A primeira delas refere-se ao fato de que basicamente todo o conhecimento atual sobre neuroproteção ocular deriva de estudos realizados em: a) animais de laboratório; b) empregando modelos de lesão neuronal que não necessariamente reproduzem as mesmas condições observadas no glaucoma; c) em culturas de células neuronais ou gliais ou d) a partir do emprego de tratamento neuroprotetor para doenças de cunho neurológico. Muitos dos mecanismos neuroquímicos envolvidos em neuroinjúria e neuroproteção podem ser específicos de partes do SNC e de algumas espécies, não estando envolvidos na lesão glaucomatosa ao nervo óptico em seres humanos. Assim sendo, o estabelecimento de modelos experimentais que reproduzam com perfeição a lesão causada pelo glaucoma em seres humanos torna-se necessário para que as potencialidades do tratamento neuroprotetor no glaucoma possam ser estabelecidas e para viabilizar estudos clínicos ulteriores.
Um segundo problema, conseqüência direta do que foi mencionado, são os efeitos dúbios provocados por certas drogas. De fato, algumas drogas mostraram simultaneamente efeito neuroprotetor, nulo ou mesmo deletério, conforme o modelo de estudo empregado, as doses administradas e a forma de administração do agente-candidato. Logo, a elucidação do verdadeiro efeito da droga deverá ser obtido através do refinamento dos modelos experimentais de estudo e de uma melhor compreensão sobre a biodisponibilidade e dos mecanismos de ação das drogas em teste. Um exemplo clássico dessa dificuldade consiste no óxido nítrico: o óxido nítrico demonstrou efeito neuroprotetor como neutralizador de radicais hidroxila quando liberado a partir de grupos nitrato provenientes da nitroglicerina, mas aumentou a produção desses radicais quando liberado a partir do nitroprussiato sódico(46, 58).
A terceira dificuldade a ser mencionada consiste na obtenção de drogas ao mesmo tempo eficazes e seguras, que superem os efeitos adversos observados nas drogas de primeira geração. Um exemplo ilustrativo desse problema pode ser encontrado no Selfotel, capaz de inibir a apoptose pelo bloqueio do receptor NMDA mas induzindo, adicionalmente, alterações comportamentais intoleráveis ao indivíduo.
Superadas essas dificuldades iniciais, estudos clínicos bem controlados irão determinar a validade do emprego dos neuroprotetores em glaucoma, as melhores drogas indicadas e as doses que proporcionarão o melhor índice terapêutico.
REFERÊNCIAS
1. Bowers WJ, Howard DF, Federoff HJ. Gene therapeutic strategies for neuroprotection: implications for Parkinson's disease. Exp Neurol 1997;144:58-68.
2. Koller WC. Neuroprotective therapy for Parkinson's disease. Exp Neurol 1997;144:24-8.
3. Isaacs KR, Wolpoe ME, Jacobowitz DM. Calretinin-immunoreactive dopaminergic neurons from embryonic rat mesencephalon are resistant to levodopa-induced neurotoxicity. Exp Neurol 1997;146:25-32.
4. Olanow CW, Jenner P, Brooks D. Dopamine agonists and neuroprotection in Parkinson's disease. Ann Neurol 1998;44(3 Suppl):S167-74.
5. Olanow CW. Attempts to obtain neuroprotection in Parkinson's disease. Neurology 1997;49(1 Suppl):S26-33.
6. Rodrigues MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol 1998;44(3 Suppl):S175-88.
7. Kragten E, Lalande I, Zimmermann K, et al. Glyceraldehyde-3-phosphate dehydrogenase, the putative target of the antiapoptotic compounds CGP 3466 and R-(-)-deprenyl. J Biol Chem 1998;273:5821-8.
8. Marsden CD, Olanow CW. The causes of Parkinson's disease are being unraveled and rational neuroprotective therapy is close to reality. Ann Neurol 1998;44 (3 Suppl)S189-96.
9. Sziraki I, Mohanakumar KP, Rauhala P, et al. Manganese: a transition metal protects nigrostriatal neurons from oxidative stress in the iron-induced animal model of parkinsonism. Neuroscience 1998;85:1101-11.
10. Ziy I, Offen D, Barzilai A et al. Modulation of control mechanisms of dopamine-induced apoptosis - a future approach to the treatment of Parkinson's disease? J Neural Transm 1997;49(Suppl):195-202.
11. Ongini E, Adami M, Ferri C, Bertorelli R. Adenosine A2A receptors and neuroprotection. Ann N Y Acad Sci 1997;825:30-48.
12. Perez-Pinzon MA, Yenari MA, Sun GH, Kunis DM, Steinberg GK. SNX-111, a novel, presynaptic N-type calcium channel antagonist, is neuroprotective against focal cerebral ischemia in rabbits. J Neurol Sci 1997;153:25-31.
13. Toung TJ, Traystman RJ, Hum PD. Estrogen-mediated neuroprotection after experimental stroke in male rats. Stroke 1998;29:1666-70.
14. Krupinski J, Issa R, Bujny T, et al. A putative role for platelet-derived growth factor in angiogenesis and neuroprotection after ischemic stroke in humans. Stroke 1997;28:564-73.
15. Lekieffre D, Benavides J, Scatton B, Nowicki JP. Neuroprotection afforded by a combination of eliprodil and a thrombolytic agent, rt-PA, in a rat thromboembolic stroke model. Brain Res 1997;776:88-95.
16. Rudolphi KA, Schubert P. Modulation of neuronal and glial cell function by adenosine and neuroprotection in vascular dementia. Behav Brain Res 1997;83:123-8.
17. Ma J, Endres M, Moskowitz MA. Synergistic effects of caspase inhibitors and MK-801 in brain injury after transient focal cerebral ischaemia in mice. Br J Pharmacol 1998;124:756-62.
18. Bertorelli R, Adami M, Di Santo E, Ghezzi P. MK 801 and dexamethasone reduce both tumor necrosis factor levels and infarct volume after focal cerebral ischemia in the rat brain. Neurosci Lett 1998;246:41-4.
19. Tsuchida E, Rice M, Bullock R. The neuroprotective effect of the forebrain-selective NMDA antagonist CP101,606 upon focal ischemic brain damage caused by acute subdural hematoma in the rat. J Neurotrauma 1997;14:409-17.
20. Behl C, Trapp T, Skutella T, Holsboer F. Protection against oxidative stress-induced neuronal cell death - a novel role for RU486. Eur J Neurosci 1997;9:912-20.
21. Behl C, Holsboer F. Oxidative stress in the pathogenesis of Alzheimer's disease and antioxidant neuroprotection. Fortschr Neurol Psychiatr 1998; 66:113-21.
22. Heidrich A, Rösler M, Riederer P. Pharmacotherapy of Alzheimer dementia: therapy of cognitive symptoms - new results of clinical studies. Fortschr Neurol Psychiatr 1997;65:108-21.
23. Yoles E, Schwartz M. Potencial neuroprotective therapy for glaucomatous optic neuropathy. Surv Ophthalmol 1998;42:367-372.
24. Dreyer EB, Grosskreutz CL. Excitatory mechanisms in retinal ganglion cell death in primary open angle glaucoma (POAG). Clin Neurosci 1997;4:270-3.
25. Schwartz M, Belkin M, Yoles E, Solomon A. Potential treatment modalities for glaucomatous neuropathy: neuroprotection and neuroregeneration. J Glaucoma 1996;5:427-32.
26. Chew SJ, Ritch R. Neuroprotection: the next breakthrough in glaucoma? Proceedings of the Third Annual Optic Nerve Rescue and Restoration Think Tank. J Glaucoma 1997;6:263-6.
27. Brubaker RF. Delayed functional loss in glaucoma. LII Edward Jackson Memorial Lecture. Am J Ophthalmol 1996;121:473-83.
28. Anderson RS, O'Brien C. Psychophysical evidence for a selective loss of M ganglion cells in glaucoma. Vision Res 1997;37:1079-83.
29. Porciatti V, Di Bartolo E, Nardi N, Fiorentini A. Responses to chromatic and luminance contrast in glaucoma: a psychophysical and electrophysiological study. Vision Res 1997;37:1975-87.
30. Nickells RW. Retinal ganglion cell death in glaucoma: the how, the why, and the maybe. J Glaucoma 1996;5:345-56.
31. Perlman JI, McCole SM, Pulluru P, Chang CJ, Lam TT, Tso MO. Disturbances in the distribution of neurotransmitters in the rat retina after ischemia. Curr Eye Res 1996;15:589-96.
32. Kerrigan LA, Zack DJ, Quigley HA, Smith SD, Pease ME. TUNEL-positive ganglion cells in human primary open-angle glaucoma. Arch Ophthalmol 1997;115:1031-5.
33. Okisaka S, Murakami A, Mizukawa A, Ito J. Apoptosis in retinal ganglion cell decrease in human glaucomatous eyes. Jpn J Ophthalmol 1997;41:84-8.
34. Sucher NJ, Lipton SA, Dreyer EB. Molecular basis of glutamate toxicity in retinal ganglion cells. Vision Res 1997;37:3483-93.
35. Kitano S, Morgan J, Caprioli J. Hypoxic and excitotoxic damage to cultured rat retinal ganglion cells. Exp Eye Res 1996;63:105-12.
36. Caprioli J, Kitano S, Morgan JE. Hyperthermia and hypoxia increase tolerance of retinal ganglion cells to anoxia and excitotoxicity. Invest Ophthalmol Vis Sci 1996;37:2376-81.
37. Carter Dawson L, Shen F, Harwerth RS et al. Glutamine immunoreactivity in Müller cells of monkey eyes with experimental glaucoma. Exp Eye Res 1998;66:537-45.
38. Dreyer EB. A proposed role for excitotoxicity in glaucoma. J Glaucoma 1998;7:62-7.
39. Maguire G, Simko H, Weinreb RN, Ayoub G. Transport-mediated release of endogenous glutamate in the vertebrate retina. Pflugers Arch 1998;436:481-4
40. Yoles E, Schwartz M. Elevation of intraocular glutamate levels in rats with partial lesion of the optic nerve. Arch Ophthalmol 1998;116:906-10.
41. Dreyer EB, Zurakowski D, Schumer RA, Podos SM, Lipton SA. Elevated glutamate levels in the vitreous body of humans and monkeys with glaucoma. Arch Ophthalmol 1996;114:299-305.
42. Oku H, Yamaguchi H, Sugiyama T, Kojima S, Ota M, Azuma I. Retinal toxicity of nitric oxide released by administration of a nitric oxide donor in the albino rabbit. Invest Ophthalmol Vis Sci 1997;38:2540-4.
43. Vorwerk CK, Hyman BT, Miller JW, et al. The role of neuronal and endothelial nitric oxide synthase in retinal excitotoxicity. Invest Ophthalmol Vis Sci 1997;38:2038-44.
44. Neufeld AH, Hernandez MR, Gonzalez M. Nitric oxide synthase in the human glaucomatous optic nerve head. Arch Ophthalmol 1997;115:497-503.
45. Farinelli SE, Greene LA, Friedman WJ. Neuroprotective actions of dipyridamole on cultured CNS neurons. J Neurosci 1998;18:5112-23.
46. Rauhala P, Khaldi A, Mohanakumar KP, Chiueh CC. Apparent role of hydroxyl radicals in oxidative brain injury induced by sodium nitroprusside. Free Radic Biol Med 1998;24:1065-73.
47. Sun F, Zhang X, Bian X. An ultrastructural study on human retina with late glaucoma. Chung Hua Yen Ko Tsa Chih 1996;32:35-7.
48. Laquis S, Chaudhary P, Sharma SC. The patterns of retinal ganglion cell death in hypertensive eyes. Brain Res 1998;784:100-4.
49. Janssen P, Naskar R, Moore S, Thanos S, Thiel HJ. Evidence for glaucoma-induced horizontal cell alterations in the human retina. Ger J Ophthalmol 1996;5:378-85.
50. Manni G, Lambiase A, Centofanti M, et al. Histopathological evaluation of retinal damage during intraocular hypertension in rabbit: involvement of ganglion cells and nerve fiber layer. Graefes Arch Clin Exp Ophthalmol 1996;234(Suppl 1):S209-13.
51. Akaike A, Adachi K, Kaneda K. Techniques for evaluating neuronal death of the retina in vitro and in vivo. Nippon Yakurigaku Zasshi 1998;111:97-104.
52. Zurakowski D, Vorwerk CK, Gorla M, et al. Nitrate therapy may retard glaucomatous optic neuropathy, perhaps through modulation of glutamate receptors. Vision Res 1998;38:1489-94.
53. Jia WW, Wang Y, Qiang D, Tufaro F, Remington R, Cynader M. A bcl-2 expressing viral vector protects cortical neurons from excitotoxicity even when administered several hours after the toxic insult. Brain Res Mol Brain Res 1996;42:350-3.
54. Hu DN, Ritch R. Tissue culture of adult human retinal ganglion cells. J Glaucoma 1997;6:37-43.
55. Baumgartner BJ, Shine HD. Neuroprotection of spinal motoneurons following targeted transduction with an adenoviral vector carrying the gene for glial cell line-derived neurotrophic factor. Exp Neurol 1998;153:102-12.
56. Kovács AD, Szabó G. Targeted transduction of CNS neurons with adenoviral vectors carrying neurotrophic factor genes confers neuroprotection that exceeds the transduced population. J Neurosci 1997;17:6504-11.
57. Lou J, Lenke LG, Xu F, O'Brien M. In vivo Bcl-2 oncogene neuronal expression in the rat spinal cord. Spine 1998;23:517-23.
58. Mohanakumar KP, Hanbauer I, Chiueh CC. Neuroprotection by nitric oxide against hydroxyl radical- induced nigral neurotoxicity. J Chem Neuroanat 1998;14:195-205.
Endereço para correspondência:
Dr. Flávio A. Marigo
Rua dos Otoni, 881 - 13º andar - Santa Efigênia
Belo Horizonte (MG) CEP 30150-270
Tel.: (31) 274-3355 - Fax (31) 222-5083
E-mail: [email protected]
Descritores: Glaucoma/patologia; Glaucoma/quimioterapia; Apoptose; Agentes neuroprotetores/uso terapêutico
Keywords: Glaucoma/pathology; Glaucoma/drug therapy; Apoptosis; Neuroprotective agents/therapeutic use
Trabalho realizado no Serviço de Glaucoma do Hospital São Geraldo da Universidade Federal de Minas Gerais, Belo Horizonte (MG).
1 Doutor em Oftalmologia pela Universidade Federal de Minas Gerais. Pós-Doutorado, The New York Eye and Ear Infirmary, New York Medical College, USA.
2 Professor Titular do Departamento de Oftalmologia da Faculdade de Medicina da Universidade Federal de Minas Gerais.
3 Serviço de Glaucoma do Hospital São Geraldo da Universidade Federal de Minas Gerais.
4 Serviço de Glaucoma do Instituto da Visão, Minas Gerais.
Os autores não têm interesse comercial ou financeiro em nenhum dos instrumentos ou técnicas descritas nesse trabalho.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.