Arq. Bras. Oftalmol. 2009; 72 (1): 10.1590/S0004-27492009000100019
Total: 2067
Luiz Alberto Zago Filho1; Naoye Shiokawa1
DOI: 10.1590/S0004-27492009000100019
RESUMO
Descrevem-se dois casos em que os pacientes tinham como queixa principal a baixa da acuidade visual progressiva em evolução de quatro anos e oito meses, respectivamente. O quadro clínico de oftalmoplegia externa, com alterações fundoscópicas demonstrando atrofia da retina, epitélio pigmentar e coriocapilar, levou ao diagnóstico clínico de síndrome de Kearns-Sayre. A investigação etiológica do quadro oftalmológico é fundamental, pois na maioria das vezes está associado a condições sistêmicas graves que necessitam de acompanhamento e tratamento adequados.
Descritores: Doenças mitocondriais; Doenças mitocondriais; Oftalmoplegia; Ptose palpebral; Bloqueio cardíaco; Degeneração retiniana; Relatos de casos
ABSTRACT
Two cases in which patients had progressive loss of visual acuity in four years and eight months respectively are described. Clinical examination revealed fundoscopic alterations of the retina, pigment epithelium and choriocapillaris showing marked atrophy. The clinical picture resembles Kearns-Sayre syndrome. Etiological investigation is fundamental, because most of time this condition is associated with more serious systemic complications which need specific care.
Keywords: Mitochondrial diseases; Mitochondrial diseases; Ophthalmoplegia; Palpebral ptosis; Heart block; Retinal degeneration; Case reports
INTRODUÇÃO
A síndrome de Kearns-Sayre foi descrita em 1958 por ambos autores como relato de caso de dois pacientes portadores dessa disfunção, porém só em 1965 teve reconhecimento como uma nova síndrome(1).
Na maioria das vezes é sugestiva, manifestando sintomas de fraqueza muscular de membros superiores e inferiores, fadiga, ptose palpebral e baixa da acuidade visual. Na avaliação oftalmológica dos pacientes observa-se oftalmoplegia externa progressiva com notada diminuição das versões, ptose palpebral moderada a acentuada, degeneração da retina e epitélio pigmentar. Baixa estatura, sinais de disfunção cerebelar como ataxia, bloqueio de condução cardíaco sintomático, diabete mellitus, surdez, entre outras manifestações multissistêmicas também são encontradas(2).
Alterações no DNA mitocondrial (mtDNA) são implicadas como fatores causais da síndrome. A mitocôndria possui seu próprio DNA, e o defeito mais comum observado é uma ampla deleção entre 1,3 a 8 kilobases do braço do mtDNA. Diferentes anomalias na cadeia têm sido observadas, porém são consideradas expressões clínicas incompletas de Kearns-Sayre. Essa deleção colabora para o mau funcionamento da função mitocondrial, o que leva a manifestações clínicas de fadiga muscular e oftalmoplegia(3).
Os sinais iniciam-se geralmente antes dos 20 anos, com sintomas oftalmológicos, neuromusculares e cardiológicos evidenciáveis. A investigação etiológica é imprescindível já que muitos pacientes desenvolvem bloqueio de condução cardíaco súbito, assim como demência e distúrbios de equilíbrio.
RELATO DE CASO
Caso 1
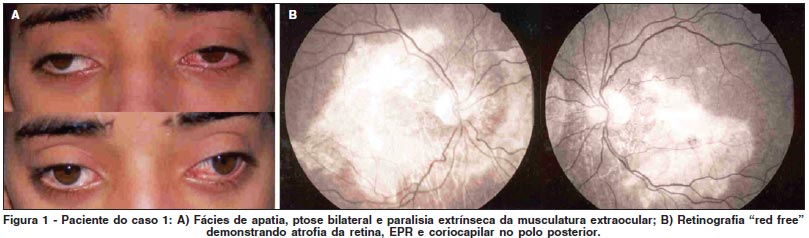
C.D.S., masculino, estudante, 15 anos, branco, natural e procedente de Boa Vista (RO), veio para avaliação com queixa de baixa de acuidade visual bilateral, progressiva há quatro anos com piora significativa no último ano. Ele não relatava outras queixas, nem sistêmicas, nem relativa a antecedentes pessoais ou familiares. No exame físico geral observou-se baixa estatura para a idade, hipotrofismo muscular generalizado, fraqueza muscular e fácies de apatia. A ectoscopia demonstrou ptose parcial bilateral moderada simétrica poupando a área pupilar. A motilidade ocular extrínseca encontrava-se gravemente diminuída em todas as posições do olhar, não apresentando qualquer grau de convergência. Na levoversão não apresentava movimentação do olho direito (Figura 1A). A acuidade visual corrigida era de 20/800 e 20/400. Os reflexos fotomotores estavam preservados. A oftalmoscopia binocular constatou extensas áreas de atrofia de retina e coriocapilar no polo posterior abrangendo a área macular, assim como intensa dispersão difusa do epitélio pigmentar da retina. A retinografia e a angiografia fluoresceínica demonstraram visualização dos grandes vasos da coróide por atrofia da retina, epitélio pigmentar e coriocapilar (Figura 1B). O paciente foi encaminhado ao serviço de cardiologia e neurologia para avaliação, onde se constatou bloqueio cardíaco completo de ramo direito e incompleto esquerdo, que levou à implantação imediata de marcapasso cardíaco. O paciente foi liberado para continuar o tratamento em seu local de origem, após alta hospitalar e orientações sobre a patologia.
Caso 2
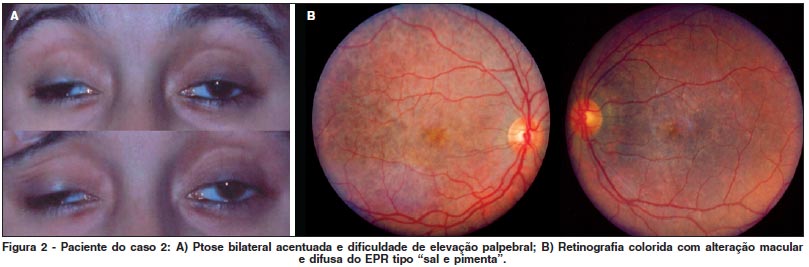
R.S.S., 30 anos, feminina, branca, natural e procedente de Laranjeiras do Sul (PR) apresentava baixa da acuidade visual havia oito meses, associada a ptose bilateral progressiva há um ano e fraqueza generalizada, sem nenhum antecedente significativo pessoal ou familiar. A ectoscopia apresentava ptose bilateral importante, diminuição da incursão de ambas pálpebras superiores, e elevação dos músculos frontais e corrugadores tentando aumentar a fenda palpebral. As fendas mediam 5 e 8 milímetros respectivamente (Figura 2A). A área pupilar do olho direito encontrava-se ocluída na posição primária, obrigando a paciente posicionar-se com o mento erguido. A motilidade extrínseca encontrava-se moderadamente diminuída principalmente nas versões horizontais e a musculatura intrínseca apresentava reflexos normais. A acuidade visual corrigida era de 20/80 e 20/100. A oftalmoscopia binocular indireta evidenciou áreas atróficas de retina e epitélio pigmentar macular limitadas, assim como dispersão pigmentar do tipo "sal e pimenta" até a periferia (Figura 2B). Essa paciente também foi encaminhada para investigação cardiológica e neurológica, sendo acompanhada sem qualquer intervenção. O bloqueio de condução parcial diagnosticado na ocasião ainda não requer intervenção. Os autores avaliam a possibilidade de operação da ptose palpebral.
DISCUSSÃO
A síndrome de Kearns-Sayre é uma doença extremamente rara, com prevalência desconhecida na literatura internacional, e escassos casos publicados no Brasil. Acredita-se que variações patológicas no mtDNA aconteçam em 1 a cada 8.000 indivíduos, nem todas traduzindo-se clinicamente(4).
As mutações no mtDNA são geralmente herdadas pela linhagem materna, entretanto mutações espontâneas podem acontecer. O tipo de repercussão clínica vai depender da quantidade de mtDNA anormal, situação conhecida como heteroplasmia(5).
A mitocôndria desempenha papel principal no metabolismo aeróbico através da fosforilação oxidativa. Este processo é conduzido pela cadeia respiratória mitocondrial com a interação de complexos enzimáticos situados na porção interna da membrana da mitocôndria. O resultado desta seqüência de eventos é a geração de adenosina trifosfato (ATP).
A ATP é uma molécula que fornece energia a todo processo celular ativo. A diminuição da sua produção, e a tentativa de gerar energia através de mecanismos anaeróbios são algumas das características comuns às patologias mitocondriais, que por fim acumulam lactase e N-aspartato(6).
Doenças mitocondriais afetam tecidos que dependem substancialmente do processo de fosforilação oxidativa. Órgãos que necessitam de grande quantidade de energia, como o tecido muscular e nervoso, células retinianas e cocleares responsáveis pela audição, são mais comumente acometidas.
O caminho que promove dano nas células retinianas não está esclarecido, apenas existe a constatação de que células humanas respondem de diferentes maneiras à mudança do seu DNA nuclear. A alteração comum à doença de Kearns-Sayre é a retinopatia em sal e pimenta periférica, com atrofia do epitélio pigmentar no polo posterior(7).
Os critérios de diagnóstico da síndrome são a tríade de oftalmoplegia progressiva, degeneração pigmentar retiniana e bloqueio de condução cardíaco. A biópsia de tecido muscular com coloração de Gomori constata mudanças morfológicas características nas fibras estriadas também chamadas de "ragged-red cells" (fibras vermelhas rasgadas)(8).
A constatação de uma larga deleção no mtDNA obtida por estudo genético com cadeia de polimerase (PCR), também permite estabelecer o diagnóstico nestes casos evitando-se biópsia.
É importante salientar a necessidade da avaliação precoce como também conhecer diagnósticos diferenciais. Algumas patologias que apresentam quadro associado a retinose pigmentar são as síndromes de Usher, Laurence-Moon, Bardet-Biedl, doença de Batten, doença de Refsum e síndrome de Sênior-Loken(9).
Miopatias inflamatórias muitas vezes confundem os quadros associados à disfunção mitocondrial e distonias neuromusculares. Outras doenças mitocondriais também cursam com disfunção muscular progressiva como a NARP (Neuropatia, ataxia, retinose pigmentar) e a ARCO (Cardiomiopatia autossômica recessiva e oftalmoplegia). Miopatias associadas à disfunção muscular ocular e periocular como a oftalmoplegia externa crônica progressiva, distrofia miotônica e miastenia grave encontram-se entre os diagnósticos diferenciais.
Não há tratamento efetivo para controle da disfunção muscular e da retinopatia. Um estudo randomizado, prospectivo, duplo-cego com 15 pacientes portadores de síndrome de Kearns-Sayre e oftalmoplegia externa crônica progressiva utilizou o monoidrato de creatina oral. Uma dosagem de 150 mg/kg versus placebo não demonstrou melhora nos sintomas clínicos musculares na série(10).
Também a coenzima Q(10) não apresentou resultados satisfatórios na disfunção muscular em um pequeno grupo de pacientes com a síndrome.
Estudos demonstram que aproximadamente 57% dos pacientes com a síndrome apresentam sinais clínicos de disfunção cardíaca. A avaliação cardiológica é de fundamental importância devido ao risco associado de bloqueio de condução e morte súbita.
Nos dois casos apresentados as manifestações comuns encontradas foram a oftalmoplegia, ptose palpebral e a retinopatia pigmentar. O segundo caso não apresentou disfunções neurológicas graves ou sintomatologia cardíaca acentuada apesar de demonstrar importantes alterações eletrocardiográficas. Demonstraram-se portanto dois casos da mesma síndrome manifestando-se em fenótipos, idade e estágios diferentes.
O avanço nas técnicas sobre os mecanismos de alteração do mtDNA e das formas de transmissão hoje são as grandes esperanças para o manejo da doença e também para o aconselhamento genético. O entendimento das características desta síndrome torna-se essencial para a investigação apropriada e manejo de suspeitos.
REFERÊNCIAS
1. Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol. 1958;60(2):280-9.
2. Glutz von Blotzheim S, Borruat FX, Hirt L. [Ocular mitochondrial myopathies: a spectrum of clinical presentations]. Klin Monatsbl Augenheilkd. 1998;212(5):299-300. French.
3. Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA, Rowland LP. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;51(6):1525 and 8 pages following.
4. Chinnery PF, DiMauro S, Shanske S, Schon EA, Zeviani M, Mariotti C, et al. Risk of developing a mitochondrial DNA deletion disorder. Lancet. 2004; 364(9434):592-6.
5. Ramírez-Miranda A, Navas-Pérez A, Gurria-Quintana L, Vargas-Ortega J, Murillo-Correa C, Zenteno JC. [PCR-based detection of heteroplasmic deleted mitochondrial DNA in Kearns-Sayre syndrome]. Arch Soc Esp Oftalmol. 2008;83(3):155-9. Spanish.
6. Johns DR. Seminars in medicine of the Beth Israel Hospital, Boston. Mitochondrial DNA and disease. N Engl J Med. 1995;333(10):638-44. Commentin: N Engl J Med. 1996;334(4):270-1. N Engl J Med. 1996;334(4):270; author reply 271.
7. Isashiki Y, Nakagawa M, Ohba N, Kamimura K, Sakoda Y, Higuchi I, et al. Retinal manifestations in mitochondrial diseases associated with mitochondrial DNA mutation. Acta Ophthalmol Scand. 1998;76(1):6-13.
8. Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989;320(20):1293-9. Comment in: N Engl J Med. 1990;322(10):701.
9. Seigel AM, Shaywitz BA, Ciesielski T. Kearns-Sayre syndrome: the importance of early recognition. Am J Dis Child. 1977;131(6):711-2.
10. Kornblum C, Schröder R, Müller K, Vorgerd M, Eggers J, Bogdanow M, et al. Creatine has no beneficial effect on skeletal muscle energy metabolism in patients with single mitochondrial DNA deletions: a placebo-controlled, double-blind 31P-MRS crossover study. Eur J Neurol. 2005;12(4):300-9.
Endereço para correspondência:
Luiz Alberto Zago Filho
Rua Nilo Peçanha, 605
Lages (SC) CEP 88523-330
Tel.: (49) 3222-2682, 3223-1450, 9986-2600
E-mail: [email protected]
Recebido para publicação em 09.07.2007
Última versão recebida em 29.07.2008
Aprovação em 30.10.2008
Trabalho realizado no Departamento de Oftalmologia e Otorrino da Universidade Federal do Paraná - UFPR - Curitiba (PR) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência do Dr. João J. Nassaralla Jr. sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}