Arq. Bras. Oftalmol. 2008; 71 (6): 10.1590/S0004-27492008000600023
Total: 2600
Aline Amaral Fulgêncio da Cunha1; Bruno Carvalho Picanço1; Grazziella Acácio e Almeida1; Nara Helena Teixeira Rodrigues1; Guilherme Mourão Soares da Rocha1
DOI: 10.1590/S0004-27492008000600023
RESUMO
Apresentação de caso clínico de paciente de 30 anos com quadro de baixa acuidade visual progressiva há 8 anos, sem diagnóstico conclusivo. Após exame oftalmológico completo, realizado na unidade de urgência oftalmológica da Clínica de Olhos da Santa Casa de Belo Horizonte, foi observado alteração macular em ambos os olhos com aspecto cistóide "em roda de carroça". A ausência de extravasamentos de contraste à angiofluoresceinografia e a presença de cistos e aumento da espessura foveal demonstrados pela tomografia de coerência óptica sugeriram tratar-se de retinosquise juvenil que pôde ser confirmada com o eletrorretinograma.
Descritores: Retinosquise; Mácula lutea; Tomografia de coerência óptica; Angiofluoresceinografia; Eletrorretinografia; Relatos de casos
ABSTRACT
We report a 30-year-old patient presenting a non-conclusive diagnosis of low progressive visual acuity for 8 years. A cystoid maculopathy (stellate striation) was observed in both eyes after a complete ophthalmologic examination performed in the emergency ward at the Clínica de Olhos da Sanata Casa de Belo Horizonte. The absence of contrast leakage in the foveal region identified by fluorescein angiography and the presence of cysts and increase of foveal thickness in optical coherence tomography suggested juvenile retinoschisis which could be confirmed through electroretinogram.
Keywords: Retinoschisis; Macula lutea; Tomography, optical coherence; Fluorescein angiography; Electroretinography; Case reports
INTRODUÇÃO
A retinosquise juvenil ou retinosquise congênita é uma distrofia vitreorretiniana bilateral, assimétrica com defeito básico nas células de Müller e caracteriza-se pela separação da camada de fibras nervosas do restante da retina sensorial(1). Trata-se de uma patologia rara, hereditária, transmitida de forma recessiva através de uma alteração no braço curto do cromossomo X, afetando quase exclusivamente o sexo masculino(2). Manifesta-se pela ocorrência de baixa acuidade visual entre 5 e 10 anos de idade, com dificuldades na leitura, e progressão nas primeiras duas décadas de vida. Em geral, a acuidade visual permanece estável, aproximadamente 20/60, após a segunda década. Em torno da 5ª e 6ª décadas de vida pode haver piora da acuidade visual devido à degeneração macular, podendo chegar a 20/200.
Possui como principais complicações descolamento de retina e hemorragia vítrea.
RELATO DO CASO
Paciente do sexo masculino, 30 anos, natural e procedente de Ibiaí - MG.
Veio encaminhado ao serviço de Urgência e Emergência da Clínica de Olhos da Santa Casa de Belo Horizonte, com relato de baixa acuidade visual em ambos os olhos (AO), de evolução progressiva há 8 anos e sem diagnóstico até o presente momento. Negava quaisquer outros sintomas oculares, como dor, hiperemia ou secreção; sem antecedentes de patologia ocular prévia. Negava ainda antecedentes mórbidos ou uso de medicamentos. Ausência de história familiar para patologias oculares (sic).
Ao exame oftalmológico apresentava acuidade visual de 20/70 no olho direito (OD) e 20/60 no olho esquerdo (OE), sem melhora ao exame refracional. Biomicroscopia e medida da pressão ocular sem alterações significativas. Ao mapeamento de retina evidenciava-se mácula com aspecto cistóide, tipo "Roda de carroça" (estrias radiais), em AO, além de degeneração tipo lattice na periferia retiniana temporal inferior do OD. Não foram observadas roturas ou outras degenerações retinianas periféricas.
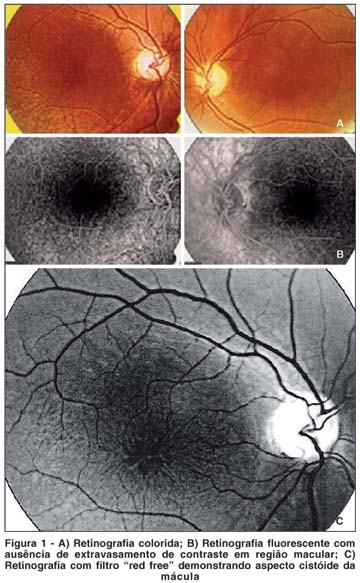
A angiofluoresceinografia não demonstrava qualquer alteração da fluorescência retinocoroidiana ou vascular. Não houve extravasamento de contraste na região macular em nenhuma fase do exame (Figura 1B).
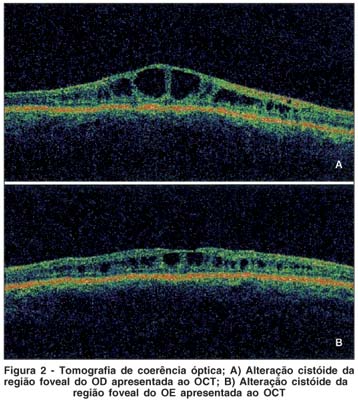
A tomografia de coerência óptica (OCT) demonstrava ausência do contorno foveolar, aplanamento e irregularidades na superfície retiniana interna pela presença de cistos subfoveais, de baixa refletividade, especialmente nas camadas externas. Espessura retiniana central: OD: 402 micra/ OE: 261 micra; espessura foveal: OD: 441 micra/ OE 275 micra (Figura 2).
Ao eletrorretinograma (ERG) apresentava onda b escotópica de amplitude diminuída além de resposta combinada em padrão negativa, com onda b menor que onda a. Respostas fotópicas de amplitude diminuída e tempo implícito no limiar da normalidade.
Teste de Ishihara sem alteração.
Teste de Farnsworth-Munsell apresentou alterações compatíveis com defeito tritan (sensibilidade diminuída para o azul).
DISCUSSÃO
A retinosquise juvenil, também chamada retinosquise hereditária congênita, é uma desordem vítreorretiniana relativamente rara, que origina baixa acuidade visual em homens jovens(1-6).
É transmitida como herança ligada ao X e tem como principal causador o gene XLRS1 e suas mutações(2-3,7-8).
A baixa acuidade visual desenvolve-se na primeira década, é bilateral, em geral com rápida progressão até os dez anos de idade. Frequentemente se torna estável em pacientes em torno de 20 anos podendo progredir em idade mais avançada(1,5-7).
No caso reportado neste artigo, a acuidade visual do paciente de 20/70 no OD e 20/60 no OE foi compatível com as acuidades visuais descritas na literatura. Piores acuidades visuais, como 20/100, 20/200 ou percepção de luz foram observadas em pacientes com complicações da retinosquise juvenil com descolamento de retina atingindo região macular, cicatrizes atróficas foveais e buracos maculares(1,7,9).
Mácula com estriações radiadas e aspecto cistóide é o achado principal da retinosquise em pacientes jovens além da retinosquise inferotemporal bilateral. No caso descrito neste artigo não foi encontrado degeneração retiniana periférica tipo retinosquise em nenhum dos olhos, por outro lado a lesão macular bilateral encontrada foi característica(1,3,5,7,9-11).
A retinografia fluorescente, neste caso descrito, teve sua contribuição no diagnóstico diferencial entre retinosquise e edema macular cistóide. Como não houve qualquer extravasamento de contrate ("leakage") na região macular, o que é característico do edema cistóide, a hipótese de retinosquise juvenil se intensificou(1,6,11).
O OCT também tem se demonstrado um importante adjuvante no diagnóstico de retinosquise juvenil. Várias alterações podem ser observadas com este exame, especialmente o aumento da espessura retiniana na região macular à custa de espaços císticos entre as camadas da retina nesta área, como foi demonstrado neste relato de caso(1,4,7,9,10,12).
Como não foi possível a realização do estudo genético do paciente descrito deste relato de caso, o ERG foi de grande valor na confirmação diagnóstica. Nos casos de retinosquise juvenil as amplitudes escotópicas são mais gravemente afetadas que as fotópicas. A onda a pode ser normal ou pouco diminuída, porém a onda b mostra-se significativamente reduzida tendo a aparência de onda negativa. Os potenciais oscilatórios também encontram-se reduzidos. O paciente descrito neste artigo apresentou todas estas alterações ao ERG o que corroborou a hipótese de retinosquise juvenil.
REFERÊNCIAS
1. Edwards Albert O, Robertson Joseph E J. X-linked juvenile retinoschisis. In: Ryan SJ, editor-in-chief. Retina. St. Louis: Mosby; 2001. p.487-90.
2. Inoue Y, Yamamoto S, Inoue T, Fujikado T, Kusaka S, Ohguro N, et al. Two novel point mutations of the XLRS1 gene in patients with X-linked juvenile retinoschisis. Am J Ophthalmol. 2002;134(4):622-4.
3. Dodds JA, Srivastava AK, Holden KR. Unusual phenotypic expression of an XLRS1 mutation in X-linked juvenile retinoschisis. J Child Neurol. 2006; 21(4):331-3.
4. Göbel W, Hartmann F, Haigis W. [Determination of retinal thickness in relation to the age and axial length using optical coherence tomography]. Ophthalmologe. 2001;98(2):157-62. German.
5. Brockhurst RJ. Photocoagulation in congenital retinoschisis. Arch Ophthalmol. 1970;84(2):158-65.
6. Constantaras AA, Dobbie JG, Choromokos EA, Frenkel M. Juvenile sex-linked recessive retinoschisis in a black family. Am J Ophthalmol. 1972; 74(6):1166-78.
7. Ikeda F, Iida T, Kishi S. Resolution of retinoschisis after vitreous surgery in X-linked retinoschisis. Ophthalmology. 2008;115(4):718-722.e1. Comment in: Ophthalmology. 2008;115(5):920-1; author reply 921.
8. Wu WC, Drenser KA, Capone A, Williams GA, Trese MT. Plasmin enzyme-assisted vitreoretinal surgery in congenital X-linked retinoschisis: surgical techniques based on a new classification system. Retina. 2007;27(8):1079-85.
9. Shukla D, Rajendran A, Gibbs D, Suganthalakshmi B, Zhang K, Sundaresan P. Unusual manifestations of x-linked retinoschisis: clinical profile and diagnostic evaluation. Am J Ophthalmol. 2007;144(3):419-23.
10. Benhamou N, Massin P, Haouchine B, Erginay A, Gaudric A. Macular retinoschisis in highly myopic eyes. Am J Ophthalmol. 2002;133(6):794-800.
11. Krause U, Vainio-Mattila B, Eriksson A, Forsius H. Fluorescein angiographic studies of x-chromosomal retinosquisis. Acta Ophthalmol (Copenh). 1970;48(4): 794-807.
12. Urrets-Zavalía JA, Venturino JP, Mercado J, Urrets-Zavalía EA. Macular and extramacular optical coherence tomography findings in X-linked retinoschisis. Ophthalmic Surg Lasers Imaging. 2007;38(5):417-22.
Endereço para correspondência:
Aline Amaral Fulgêncio da Cunha
Rua Odilon Braga, 735 - Apto. 101
Belo Horizonte (MG) CEP 30310-390
E-mail: [email protected]
Recebido para publicação em 01.10.2007
Última versão recebida em 13.05.2008
Aprovação em 05.09.2008
Trabalho realizado na Clínica de Olhos da Santa Casa de Belo Horizonte, Faculdade de Ciências Médicas de Minas Gerais - Belo Horizonte (MG) - Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.