Arq. Bras. Oftalmol. 2008; 71 (4): 10.1590/S0004-27492008000400026
Total: 1567
Luciano Sousa Pereira1; Timothy James McCulley3
DOI: 10.1590/S0004-27492008000400026
ABSTRACT
Gliomas are the most common infiltrative neoplasms of the optic nerve and can present as two distinct growth patterns: intraneural glial proliferation and perineural arachnoidal gliomatosis (PAG). It has been suggested that perineural arachnoidal gliomatosis is seen almost exclusively in the setting of neurofibromatosis type 1 (NF1). We describe a child with perineural arachnoidal gliomatosis occurring without neurofibromatosis type 1, supported by both radiographic and histological findings. A 4-year-old female without neurofibromatosis type 1 presented with rapidly progressive right-sided proptosis. Magnetic resonance imaging (MRI) revealed an enhancing fusiform intraconal lesion, which was hypointense on T1 and hyperintense on T2-weighted images: characteristic of perineural arachnoidal gliomatosis, the optic nerve was visualized coursing the tumor. Histopathologic study was consistent with perineural arachnoidal gliomatosis. Perineural arachnoidal gliomatosis can develop independent of neurofibromatosis type 1, as demonstrated by this case.
Keywords: Optic nerve glioma; Neurofibromatosis; Magnetic resonance imaging; Human; Female; Child; Case reports
RESUMO
Gliomas são as neoplasias infiltrativas mais freqüentes do nervo óptico e podem se apresentar através de dois padrões distintos de crescimento: proliferação glial intraneural e gliomatose aracnoidal perineural. Existem evidências de que a gliomatose aracnoidal perineural é vista quase exclusivamente em pacientes com neurofibromatose tipo 1. Descrevemos um caso de gliomatose aracnoidal perineural ocorrendo em criança sem neurofibromatose tipo 1, comprovado tanto por achados radiológicos quanto histológicos. Uma criança de quatro anos de idade, do sexo feminino, sem evidências de neurofibromatose tipo 1, apresentou quadro de proptose à direita rapidamente progressiva. Ressonância magnética revelou lesão intraconal fusiforme hipointensa em T1 e hiperintensa em T2 - característico de gliomatose aracnoidal perineural, o nervo óptico pôde ser observado atravessando o tumor. O estudo histológico foi consistente com gliomatose aracnoidal perineural. Gliomatose aracnoidal perineural pode se desenvolver independente da presença de neurofibromatose tipo 1, como demonstrado por esse caso.
Descritores: Glioma de nervo óptico; Neurofibromatose; Ressonância magnética; Humano; Feminino; Criança; Relatos de casos
RELATOS DE CASOS
Perineural arachnoidal gliomatosis: case report
Gliomatose aracnoidal perineural: relato de caso
Luciano Sousa PereiraI; Timothy James McCulleyII
IDepartment of Ophthalmology, University of California San Francisco - San Francisco - CA. Departament of Ophthalmology, Santa Casa de Misericórdia de São Paulo - São Paulo (SP) - Brazil
IIDepartment of Ophthalmology, Santa Casa de Misericórdia de São Paulo - São Paulo (SP) - Brazil
ABSTRACT
Gliomas are the most common infiltrative neoplasms of the optic nerve and can present as two distinct growth patterns: intraneural glial proliferation and perineural arachnoidal gliomatosis (PAG). It has been suggested that perineural arachnoidal gliomatosis is seen almost exclusively in the setting of neurofibromatosis type 1 (NF1). We describe a child with perineural arachnoidal gliomatosis occurring without neurofibromatosis type 1, supported by both radiographic and histological findings. A 4-year-old female without neurofibromatosis type 1 presented with rapidly progressive right-sided proptosis. Magnetic resonance imaging (MRI) revealed an enhancing fusiform intraconal lesion, which was hypointense on T1 and hyperintense on T2-weighted images: characteristic of perineural arachnoidal gliomatosis, the optic nerve was visualized coursing the tumor. Histopathologic study was consistent with perineural arachnoidal gliomatosis. Perineural arachnoidal gliomatosis can develop independent of neurofibromatosis type 1, as demonstrated by this case.
Keywords: Optic nerve glioma; Neurofibromatosis/pathology; Magnetic resonance imaging; Human; Female; Child; Case reports [Publication type]
RESUMO
Gliomas são as neoplasias infiltrativas mais freqüentes do nervo óptico e podem se apresentar através de dois padrões distintos de crescimento: proliferação glial intraneural e gliomatose aracnoidal perineural. Existem evidências de que a gliomatose aracnoidal perineural é vista quase exclusivamente em pacientes com neurofibromatose tipo 1. Descrevemos um caso de gliomatose aracnoidal perineural ocorrendo em criança sem neurofibromatose tipo 1, comprovado tanto por achados radiológicos quanto histológicos. Uma criança de quatro anos de idade, do sexo feminino, sem evidências de neurofibromatose tipo 1, apresentou quadro de proptose à direita rapidamente progressiva. Ressonância magnética revelou lesão intraconal fusiforme hipointensa em T1 e hiperintensa em T2 - característico de gliomatose aracnoidal perineural, o nervo óptico pôde ser observado atravessando o tumor. O estudo histológico foi consistente com gliomatose aracnoidal perineural. Gliomatose aracnoidal perineural pode se desenvolver independente da presença de neurofibromatose tipo 1, como demonstrado por esse caso.
Descritores: Glioma de nervo óptico; Neurofibromatose/patologia; Ressonância magnética; Humano; Feminino; Criança; Relatos de casos [Tipo de publicação]
INTRODUCTION
Gliomas are the most common infiltrative neoplasms of the optic nerve accounting for 65% of such tumors, the great majority presenting as histologically benign lesions(1). Benign gliomas may adopt one of two distinct growth patterns. Most familiar is the classic intraneural glial proliferation where neoplastic growth occurs within individual fascicles. Contrastingly, perineural arachnoidal gliomatosis (PAG) is characterized by florid invasion of the leptomeninges, with relative sparing of the nerve itself. It has been suggested that the PAG is seen almost exclusively in the setting of neurofibromatosis type 1 (NF1)(2-5). In this report we describe an atypical case of biopsy proven PAG occurring in a patient lacking evidence of NF1.
CASE REPORT
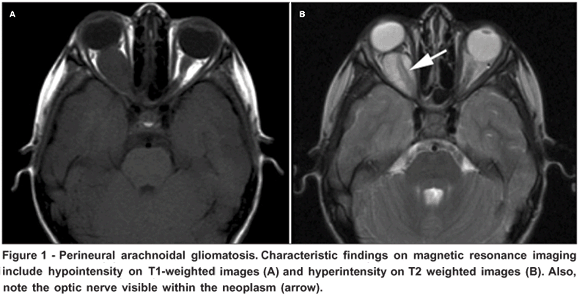
An otherwise healthy 4-year-old Hispanic girl presented with a 3 months history of rapidly progressive, painless, right-sided proptosis. On presentation she had visual acuity of 20/40 in the involved right eye with a subtle (<0.3 log unit) relative afferent pupillary defect. She had 3 mm of relative proptosis with several millimeters of inferior scleral show. Abduction, adduction and infraduction were slightly limited; supraduction was preserved. The rest of the ophthalmic examination, including anterior and posterior segment evaluations, was normal. Left eye examination was unremarkable with visual acuity recordable at 20/20. General physical examination was also normal without any findings suggestive of NF1. Magnetic resonance imaging (MRI) revealed a discrete fusiform intraconal lesion. The lesion enhanced moderately with contrast, was slightly hypointense on T1-weighted images and hyperintense on T2-weighted images (Figure 1). Slight kinking of the optic nerve was appreciable in the sagital plane. Characteristic of PAG, the nerve could be visualized coursing the tumor (Figure 1 B).
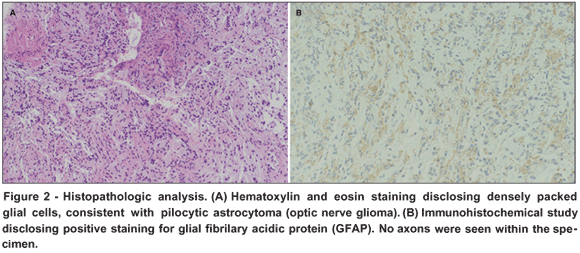
Given the rapidity of progression an incisional biopsy was performed via a lateral canthal incision. Histopathologic analysis disclosed densely packed glial cells, consistent with pilocytic astrocytoma (optic nerve glioma) (Figure 2 A). Reticulin stain highlighted neoplastic cells infiltrating the meninges, which abutted but did not infiltrate the dura. Additionally, immunohistochemical staining was positive for glial fibrillary acidic protein (GFAP) (Figure 2 B). No axons were seen within the specimen. This histologic appearance of glial cells infiltrating the meninges associated with relative sparing the optic nerve, evidenced by preservation of vision and MRI findings, is consistent with PAG.
Over the following month the degree of proptosis progressed by 4 mm and visual acuity deterioration from 20/40 to 20/60 was observed, prompting multidisciplinary intervention. Continued observation, chemotherapy, radiotherapy and surgical debulking were entertained. After much deliberation, chemotherapy (vincristine/carboplatin) was attempted without an appreciable response: proptosis progressed, however without a measurable depreciation in vision. Radiotherapy (5040 cGy) was then introduced with some success. After the third cycle visual acuity returned to 20/40 and there was a 2 mm reduction in proptosis.
DISCUSSION
The term perineural arachnoidal gliomatosis (PAG) was introduced by Stern et al. in 1980 following an assessment of histologic specimens harvested from 34 patients with optic nerve gliomas(2). The authors described two distinct growth patterns: 1) intraneural glial proliferation, expanding the individual fascicles and overall dimension of the nerve and 2) perineural arachnoidal gliomatosis (PAG), featuring tumor proliferation most predominant in the subarachnoid space. They also noted a striking association between PAG and NF1. Sixteen of 18 patients with clinical characteristics of NF1 had PAG contrasting the 16 patients without NF1, among whom only two exhibited PAG.
Subsequent radiographic studies have been consistent with the observation of PAG occurring most often in the setting of NF1(5). The first description of the magnetic resonance imaging (MRI) appearance of PAG was in 1986(4). In this report, the patient also had clinical evidence consistent with NF1. In 2001, in a retrospective review on MRI studies of 91 patients with gliomas (47 with and 44 without NF1), the optic nerve was visible coursing through the tumor in 91% (n=43) of patients with NF1 compared to 27% (n=12) without NF1(5). Despite these reports there has been a relative lack in the ophthalmic literature of PAG occurring outside the setting of NF1. Moreover, there is an overall paucity in the literature of cases with both radiographic and histologic confirmation/correlation.
Familiarity with the characteristic appearance of PAG on MRI may in many cases allow an accurate diagnosis. PAG invariably contains microcystic mucinous degeneration with a high water content yielding long T1 and T2 relaxation times. Thus PAG appears bright on T2 and dark on T1 weighted images(2,4,6-7). MRI displays a double-intensity signal characterized by a circumferential area of CSF-intensity tissue (the neoplasm itself) surrounding and sharply delineating a central linear core (the optic nerve) of opposite signal intensity: this has been termed the "pseudo-CSF" signal(6). An additional characteristic of PAG which can be appreciated radiographically is the "kinking" of optic nerve(7).
The neoplasm that PAG is most likely to be mistaken for is a meningioma. Relative preservation of the optic nerve seen with PAG with the resulting parallel borders signal can resemble "tram tracking" of meningiomas. True tram tracks of meningiomas are comprised of calcification and are most readily demonstrated by computed tomography. Menigiomas also tend to enhance intensely as compared to PAG, which exhibits only mild to moderate enhancement. If not distinguishable radiographically, the clinical setting usually differs. Gliomas occur primarily in children, often in the setting of NF1. Contrastingly, meningiomas occur most often in middle-aged women(8). Meningiomas are also seen with increased frequency in NF1 patients; however, most occur independently. Lymphoprolife rative disease is the third most likely neoplastic process to involve the optic nerve and is also worthy of consideration. Other neoplasms intrinsic to the optic nerve to consider, albeit exceedingly rare, include medulloepithelioma, ganglioglioma, hemangiopericytoma, hemangioblastoma(8).
The treatment of optic nerve glioma is controversial, with no established effective therapy(8). Inconsistent anecdotal success has been described for both radiation and chemotherapy(9-10). Surgical resection is effective however uniformly sacrifices any remaining vision and is therefore most often reserved for non-seeing eyes with disfiguring proptosis. Resection may also be indicated in patients with progressively enlarging lesion that threaten the optic chiasm or hypothalamic extension. Given the lack of response to chemotherapy and radiation and the destructive nature of resection, observation is often the most appropriate management option. The rapidity of progression in our patient prompted biopsy and subsequently chemotherapy and radiation. No appreciable response to chemotherapy was observed. Some clinical improvement was seen with radiation; however, it cannot be stated with certainty that this was not simply the natural course of the tumor and independent of intervention. Many tumors stabilize and some have been noted to regress spontaneously.
In closing, PAG is an established distinct subtype of glioma, which occurs most often in the setting of neurofibromatosis type 1. However, as demonstrated by this case PAG can develop independent of NFI. In this report we describe a child with PAG occurring without NF1, supported with both radiographic and histological findings.
REFERENCES
1. Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol. 1994; 38(5):427-52.
2. Stern J, Jakobiec FA, Housepian EM. The architecture of optic nerve gliomas with and without neurofibromatosis. Arch Ophthalmol. 1980;98(3):505-11.
3. Marquardt MD, Zimmerman LE. Histopathology of meningiomas and gliomas of the optic nerve. Hum Pathol. 1982;13(3):226-35.
4. Seiff SR, Brodsky MC, MacDonald G, Berg BO, Howes EL Jr, Hoyt WF. Orbital optic glioma in neurofibromatosis. Magnetic resonance diagnosis of perineural arachnoidal gliomatosis. Arch Ophthalmol. 1987;105(12):1689-92.
5. Korneich L, Blaser S, Schwartz M, Shuper A, Vishne T, Cohen I, et al. Optic pathway glioma: correlation of imaging findings with the presence of neurofibromatosis. AJNR Am J Neuroradiol. 2001;22(10):1963-9.
6. Brodsky MC. The "pseudo-CSF" signal of orbital optic glioma on magnetic resonance imaging a signature of neurofibromatosis. Survey Ophthalmol. 1993;38(2):213-8. Erratum in: Surv Ophthalmol. 1993;38(3):322.
7. Imes RK, Hoyt WF. Magnetic resonance imaging signs of optic nerve gliomas in neurofibromatosis 1. Am J Ophthalmol. 1991;111(6):729-34.
8. Miller NR. Primary tumors of the optic nerve and its sheath. Eye. 2004; 18(11):1026-37.
9. Grabenbauer GG, Schuchardt U, Buchfelder M, Rödel CM, Gusek G, Marx M, et al. Radiation therapy of optico-hypothalamic gliomas (OHG) - radiographic response, vision and late toxicity. Radiother Oncol. 2000;54(3): 239-45.
10. Packer RJ, Ater J, Allen J, Phillips P, Geyer R, Nicholson HS, et al. Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg. 1997;86(5):747-54.
 Correspondence address to:
Correspondence address to:
Luciano Sousa Pereira
University of California San Francisco - Department of Ophthalmology
San Francisco - CA
E-mail: [email protected]
Recebido para publicação em 28.06.2007
Última versão recebida em 31.01.2008
Aprovação em 18.05.2008
Trabalho realizado no Department of Ophthalmology - University of California - San Francisco - UCSF.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.