Arq. Bras. Oftalmol. 2008; 71 (1): 10.1590/S0004-27492008000100026
Total: 2930
Adriano de Carvalho Roma1; Paula Resende Aquino de Assis Pereira2; Adalmir Morterá Dantas3
DOI: 10.1590/S0004-27492008000100026
RESUMO
Os autores descrevem pela primeira vez no país um caso de criança do sexo feminino, com 10 anos de idade, atendida no ambulatório de Oftalmologia do Hospital Universitário Clementino Fraga Filho - UFRJ, portadora da síndrome de Leigh, que faz parte de um grupo de enfermidades metabólicas conhecidas como encefalomiopatias mitocondriais. É doença hereditária transmitida por diferentes modos de herança: mitocondrial, autossômica recessiva e recessiva ligada ao X. O início das manifestações clínicas é variado, ocorrendo em geral, dentro dos primeiros dois anos de vida, com evolução insidiosa, progressiva e com períodos de exacerbações. O diagnóstico é difícil pelo pleomorfismo de sua apresentação, sendo baseado nos achados clínicos e estudos complementares relacionados à deficiência na produção mitocondrial de ATP e da citocromo C oxidase. Como não há tratamento específico, este é baseado em medidas paliativas, portanto a identificação desta síndrome é importante como diagnóstico correto permitindo condutas adequadas à melhor qualidade de vida de seus portadores.
Descritores: Doença de Leigh; Encefalomiopatias mitocondriais; Doenças metabólicas; Mitocôndria; Citocromos C; Relatos de casos
ABSTRACT
The authors describe for the first time in the Country a case of a 10-year-old female child, assisted at the Ophthalmology Clinic of the Hospital Universitário Clementino Fraga Filho UFRJ, with Leigh's syndrome that is part of a metabolic disease group known as mitochondrial encephalomyopathies. It is an hereditary disease transmitted by a different mode of inheritance: mitochondrial, X-linked recessive and autosomal recessive. The beginning of clinical manifestations is varied and occurs usually in the first two years of life, with progressive and insidious evolution and exacerbation periods. Diagnosis is difficult because pleomorphic presentation, based on clinical findings and complementary study related to mitochondrial production of ATP and cytochrome c oxidase deficiencies. Considering that there is no specific treatment, this is based on a palliative procedure. So, the identification of this syndrome is very important to keep it under control, since its evolution is progressive.
Keywords: Leigh disease; Mitochondrial encephalomyopathies; Metabolic diseases; Mitochondria; Cytochromes c; Case reports
INTRODUÇÃO
A síndrome de Leigh também é conhecida como encefalomielopatia necrosante subaguda, encefalopatia necrosante de Leigh e encefalomielopatia necrosante de Leigh. É uma doença rara, que foi descrita por Denis Leigh em 1951 (departamento de neuropatologia do Instituto de Psiquiatria, Maudsley Hospital em Londres)(1). É uma enfermidade neurometabólica congênita, que faz parte do grupo das encefalopatias mitocondriais. Sabe-se que a alteração ocorre no metabolismo energético, sendo a principal causa de defeito na fosforilação oxidativa e geração de ATP celular(2-3). Existem três tipos de transmissão genética associada a esta síndrome: herança recessiva ligada ao X, mitocondrial e autossômica recessiva(4).
A idade de início desta doença é variada e ocorre em geral nos primeiros dois anos de vida, podendo ocorrer manifestações no adulto jovem(5). A evolução em geral é insidiosa e progressiva. O início dos sinais e sintomas ocorre de forma subaguda ou abrupta, podendo em alguns casos ser precipitado por episódios febris e por procedimentos cirúrgicos(6).
O quadro clínico caracteriza-se em crianças menores de um ano de idade com perda do controle da cabeça, hipotonia, deficiência de sugar, anorexia, vômitos, irritabilidade e convulsões. Após o primeiro ano de vida, ocorre dificuldade na marcha, ataxia, disartria, regressão intelectual, distúrbios da respiração (risco de hiperventilação ou apnéia), alterações oftalmológicas como: oftalmoplegia, nistagmo, atrofia óptica e estrabismo(4,6).
A duração da doença nos casos infantis é, em média, de um ano e nos casos tardios ou juvenis pode prolongar-se por anos(4).
As alterações histopatológicas consistem em focos bilaterais simétricos de necrose espongiforme com degeneração de mielina, proliferação vascular e gliose. A localização se dá nos núcleos da base, tálamo, tronco cerebral e medula espinhal. A tomografia computadorizada (TC) de crânio permite confirmar o diagnóstico quando se evidenciam imagens hipodensas nos núcleo da base e a ressonância nuclear magnética (RNM) quando mostra lesões menores, inclusive no tronco cerebral(4).
Exames laboratoriais que contenham os seguintes parâmetros: hiperproteinorraquia, níveis elevados de lactato e piruvato no sangue, razão lactato/piruvato no sangue e líquor elevada e a hiperlactacidemia provocada por sobrecarga glicídica, são sugestivos desta síndrome(4,6-7).
Ainda não há tratamento específico para esta patologia(4,8). Este relato foi proposto com a finalidade de descrever um caso clínico muito raro e de difícil diagnóstico, sendo sua identificação de fundamental importância para se realizar o aconselhamento genético e documentar as características clínicas da mesma e comparar os achados a outros relatos da literatura.
Caso clínico
N.V.P., 10 anos, branca, natural do Rio de Janeiro. Encaminhada ao serviço de oftalmologia do Hospital Universitário Clementino Fraga Filho - UFRJ pelo departamento de neuropediatria.
A paciente desenvolveu-se normalmente até a idade de 1 ano e 3 meses. A partir daí, começou a involuir seu desenvolvimento, não apresentando ganhos psico-motores.
Mãe relatava que a menor mostrava-se com regressão do aprendizado motor e intelectual, além de crises convulsivas e alterações do sono.
Nega história familiar positiva, irmão mais velho normal.
O exame neurológico mostrou espasticidade dos quatros membros, hipotonia muscular grave, com flexão mantida coxofemoral e joelho em extensão. Predomínio de movimentos de coreoatetose e distonia. Reflexo profundo com hipertonia de membros inferiores e reflexo superficial cutâneo plantar em extensão bilateralmente. Paralisia do IX e X pares cranianos (Figura 1).
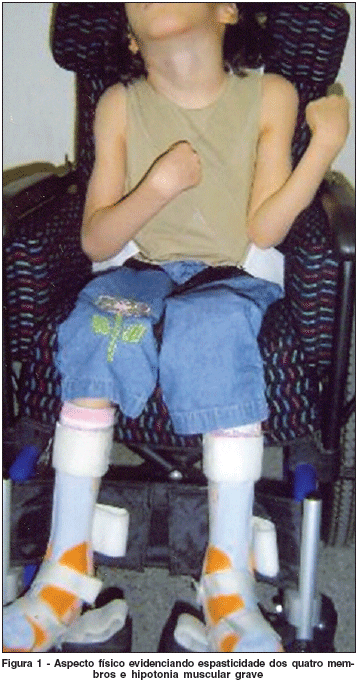
O exame de RNM constatou lesão em hipersinal em T2 periventricular, periaquedutal e de gânglios da base, e lesão em hiposinal em T1 não captante do meio de contraste, comprometendo os núcleos lenticulares e globo pálido bilateralmente com aspecto simétrico.
A TC evidenciou desmielinização e atrofia cortical.
Aminograma plasmático: aumento dos níveis de amônia, cistina, ornitina, asparagina, citrulina e glutamina.
Aminoacidúria e aumento dos níveis de lactato e piruvato no sangue.
Exame Oftalmológico:
• à ectoscopia constatou-se pequeno grau de nistagmo, sem outras alterações;
• motilidade ocular preservada, acompanhando a luz;
• reflexo fotomotor e consensual, preservados;
• acuidade visual impossibilitada de avaliação pela não cooperação da paciente;
• biomicroscopia normal em ambos os olhos;
• pressão intra-ocular normal ao toque bidigital em ambos os olhos;
• oftalmoscopia direta e indireta mostrou discos ópticos pálidos de contornos nítidos e bordos regulares, diminuição do brilho foveolar, coroidose, leve diminuição do calibre vascular, sem outras alterações (Figura 2).
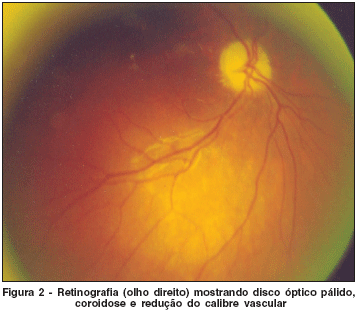
Foi realizado o potencial evocado visual (PEV) nesta paciente, que apresentou aumento da latência da onda P100 e amplitude diminuída, confirmando o diagnóstico de atrofia óptica.
DISCUSSÃO
Múltiplos defeitos genéticos, como pontos de mutação envolvendo a transcrição T-G e T-C na posição 8993 ou transição A-G na posição 8344, têm sido descritos associados a esta síndrome, provocando alteração do complexo piruvato desidrogenase, assim como dos cinco complexos que constituem a cadeia respiratória, sendo que a deficiência do citocromo C oxidase (complexo IV), provocada por mutações tanto de genes mitocondriais quanto nuclear, é o mais freqüentemente afetado nesta síndrome, sendo que a transmissão genética relacionada com o DNA mitocondrial é a mais comum(2-3,9-10).
Os achados clínicos mais freqüentes relacionados à deficiência na produção mitocondrial de ATP são a distonia e coreoatetose, e os relacionados com a deficiência do citocromo C oxidase são a ataxia e descoordenação motora, hipotonia, acidúria orgânica, movimentos anormais de membros, atraso mental e psicomotor, e com menos freqüência ocorrem a hemiplegia, hemiparesia, convulsões e epilepsias além de alterações cardíacas(4,6). No caso acima descrito, a paciente apresentava-se com essas alterações com exceção de alterações cardíacas.
Os sinais e sintomas neurológicos mais específicos são as supressões dos reflexos tendinosos com sinal de Babinski e hiperventilação intermitente(4,6). A paciente tinha no exame reflexo cutâneo plantar superficial em extensão bilateral.
Os achados oftalmológicos mais comuns são o nistagmo, movimentos oculares peculiares e menos freqüentemente temos a oftalmoplegia e a atrofia óptica, que quando ocorre é precocemente(4,6). A menor se apresentou ao exame oftalmológico com nistagmo e, embora menos comum, atrofia óptica confirmada pelo PEV. Não mostrava oftalmoplegia ou estrabismo, evidenciando a necessidade de estarmos atentos as diferentes formas de manifestações oculares nesta síndrome.
As doenças mitocondriais são diagnosticadas primariamente pela apresentação clínica, com oftalmoplegia progressiva, intolerância ao exercício com debilidade em repouso e sintomas de alterações do sistema nervoso central com miopatia associada, e, através de estudo de exames complementares, tais como a biópsia de músculo que evidencia aumento do número de mitocôndria, sendo que a histoquímica do músculo é normal, através dos níveis de CPK que podem ser normais ou ligeiramente elevados, aumento dos níveis de lactato no sangue e líquor, medição de ácido lático sérico que está aumentado, deficiência de piruvato descarboxilase e com uma relação lactato/piruvato maior que 20, sendo que na síndrome de Leigh, a presença de níveis normais não exclui o diagnóstico(4,6-7).
O diagnóstico clínico é dificultado pelo pleomorfismo de sua apresentação(4,6).
Neste caso, o diagnóstico de doença mitocondrial foi sugerido, inicialmente, pelas manifestações clínicas, e firmado com os exames complementares, como a TC de crânio e a RNM evidenciando imagens características desta síndrome, além do aumento dos níveis de lactato e piruvato no sangue e do resultado do aminograma plasmático.
Por se tratar de uma doença sem cura, o tratamento consiste em medidas paliativas com medicações anticonvulsivantes, controle da disfunção endócrina, controle dietético e procedimentos cirúrgicos caso necessário. A remoção de metabólitos nocivos objetiva o combate da acidose lática. Exercícios aeróbicos aumentam a tolerância a outros exercícios, o aconselhamento genético e o diagnóstico pré-natal têm aumentado de importância neste tipo de desordem(4,8).
O acompanhamento multidisciplinar, com a pediatria, neurologia, cardiologia, fisioterapia, oftalmologia e outras áreas, para a avaliação da gravidade das alterações e sua evolução é de suma importância, a fim de averiguar se a doença está sob controle, já que seu curso é progressivo(4,8).
REFERÊNCIAS
1. Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14(3):216-21.
2. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348(26):2656-68. Review.
3. Plaitakis A, Whetsell WO Jr, Cooper JR, Yahr MD. Chronic Leigh disease: a genetic and biochemical study. Ann Neurol. 1980;7(4):304-10.
4. Schmultzler KMRS, Pereira AMD, Vilanova LCP, Gabbai AA, Lima JGC. Encefalopatias crônicas progressivas. In: Carvalho ES, Carvalho WB, organizadores. Terapêutica e prática pediátrica. 2ª ed. São Paulo: Atheneu; 2000. p.1690-9.
5. Huntsman RJ, Sinclair DB, Bhargava R, Chan A. Atypical presentations of Leigh syndrome: a case series and review. Pediatr Neurol. 2005;32(5):334-40. Review.
6. Adams RD, Victor M, Ropper AH. Neurologia. 6ª ed. Rio de Janeiro: McGraw-Hill; 1998. p.612-53.
7. Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39(3):343-51.
8. Dimauro S, Mancuso M, Naini A. Mitochondrial encephalomyopathies: therapeutic approach. Ann N Y Acad Sci. 2004;1011:232-45.
9. Miranda AF, Ishii S, DiMauro S, Shay JW. Cytochrome C oxidase deficiency in Leigh's syndrome: genetic evidence for a nuclear DNA-encoded mutation. Neurology. 1989;39(5):697-702.
10. Kretzschmar HA, DeArmond SJ, Koch TK, Patel MS, Newth CJ, Schmidt KA, Packman S. Pyruvate dehydrogenase complex deficiency as a cause of subacute necrotizing encephalopathy (Leigh disease). Pediatrics. 1987;79(3): 370-3.
Endereço para correspondência:
Adriano de Carvalho Roma
Rua Lopes Trovão, 134, Apto. 1.102
Niterói (RJ) CEP 24220-071
E-mail: roma@ nitnet.com.br
Recebido para publicação em 21.07.2005
Última versão recebida em 29.09.2007
Aprovação em 19.10.2007
Trabalho realizado no Departamento de Oftalmologia, Hospital Universitário Clementino Fraga Filho - Universidade Federal do Rio de Janeiro - UFRJ - Rio de Janeiro (RJ) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Clélia Maria Erwenne sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.