Arq. Bras. Oftalmol. 2007; 70 (6): 10.1590/S0004-27492007000600024
Total: 4613
Ivana Cardoso Pereira1; Leandro Cabral Zacharias2; Roberta Zagui3; Ruth Santo4; Suzana Matayoshi5
DOI: 10.1590/S0004-27492007000600024
RESUMO
A granulomatose de Wegener é descrita como uma tríade de lesões: granuloma necrosante do trato respiratório, vasculite disseminada e glomerulonefrite. Pode ocorrer de maneira sistêmica ou localizada. O envolvimento ocular e orbitário é comum em ambas as formas da doença, estando presente em 50% dos casos. O exame anatomopatológico e o c-ANCA+ foram fundamentais no diagnóstico efetivo da granulomatose de Wegener a despeito do envolvimento sistêmico nos casos apresentados.
Descritores: Granulomatose de Wegener; Granulomatose de Wegener; Vasculite; Anticorpos anticitoplasma de neutrófilos; Granuloma de células plasmáticas orbital; Imunossupressores; Relatos de casos
ABSTRACT
Wegener granulomatosis (WG) is characterized by a classic triad of granulomatous inflammation of the respiratory tract, necrotizing vasculitis and nephritis. The absence of renal disease defines a subset of " limited WG" . Approximately 50% of WG patients develop ophthalmic disease. The histopatological study and +c-ANCA were essential to make a definite diagnosis in these cases.
Keywords: Wegener granulomatosis; Wegener granulomatosis; Vasculitis; Antibodies, antineutrophil cytoplasmic; Granuloma, plasma cell, orbital; Immunosuppresive agents; Case reports
INTRODUÇÃO
A granulomatose de Wegener (GW) é doença usualmente descrita como uma tríade de lesões: granuloma necrosante do trato respiratório, vasculite disseminada de arteríolas e vênulas de médio calibre e glomerulonefrite. É uma doença incomum cuja incidência verdadeira é difícil de determinar, podendo ocorrer de maneira sistêmica ou localizada. Sua forma típica caracteriza-se por ser multissistêmica com envolvimento de trato respiratório superior e inferior, seguido por falência renal devido à glomerulonefrite(1-2). A forma limitada da doença poupa os rins(3), e pode ter curso remitente com manifestações nasais, de orelha e faringe, além do acometimento pulmonar(4). O envolvimento ocular e orbitário é comum em ambas as formas da doença, estando presente em 50% dos casos(5-7), entretanto, pouco conhecida entre os oftalmologistas.
O objetivo desta apresentação é relatar quatro casos de granulomatose de Wegener, mostrando diferentes manifestações clínicas e salientando as dificuldades em se firmar o diagnóstico desta afecção em casos insidiosos e clinicamente oligossintomáticos.
RELATOS DOS CASOS
Caso 1
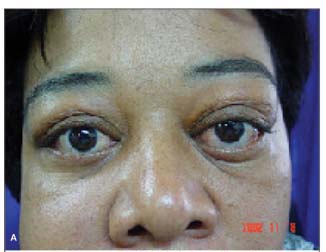
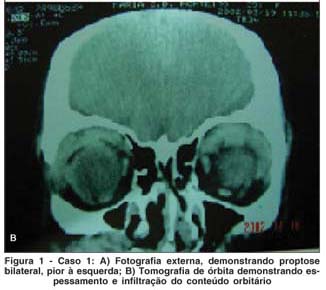
MCSM, 40 anos, sexo feminino, natural e procedente de São Paulo, aposentada. Queixa de " olhos saltados" há 5 anos. História pregressa: paciente acompanhada no departamento de Otorrinolaringologia por sinusite crônica, tendo sido submetida a 4 cirurgias em seios da face entre 1993 e 1997. Em 97, iniciou quadro de proptose bilateral, pior à esquerda, sendo feita hipótese diagnóstica de pseudotumor orbitário (Figura 1A). Foi tratada com prednisona em doses de até 80 mg/dia, com melhora parcial do quadro, mas com recidiva com a suspensão da medicação. Fez tratamento radioterápico em janeiro de 2000, apresentando melhora parcial do quadro. No período de acompanhamento, a paciente submeteu-se a uma série de biópsias para elucidação diagnóstica (incluindo biópsias de glândula lacrimal, pele palpebral e mucosa de seio maxilar) sem que se houvesse concluído a etiologia da doença. Há 18 meses, apresentava piora de edema em região maxilar direita. Há 15 meses, queixava-se de aumento da região submandibular. Exame oftalmológico: Proptose bilateral, maior à esquerda; Edema frio e xantelasma em pálpebra inferior de ambos os olhos; Motricidade ocular extrínseca e reflexos fotomotores sem alterações. Acuidade visual corrigida de 20/20 em ambos os olhos (Refração estática OD -3,00 DE; OE - 2,50 DE); Biomicroscopia: Ceratite puntata inferior leve em ambos os olhos. Pinguécula nasal em OE; Fundoscopia sem alterações em AO. Exames complementares: Tomografia de órbita com espessamento difuso da musculatura ocular extrínseca e aumento do volume de glândula lacrimal à esquerda (Figura 1A); Tomografia e ressonância de seios da face mostrando espessamento de mucosa em seio maxilar (Figura 1B); Ultra-sonografia de região cervical com sinais sugestivos de sialoadenite crônica das glândulas submandibulares e sublinguais. Linfonodos parotídeos aumentados. Pesquisa de anticorpos anti-citoplasma de neutrófilos (c-ANCA): positivo.
Baseado na clínica da paciente e no resultado do c-ANCA, foi solicitado junto ao departamento de Patologia a revisão das biópsias, cujo resultado mostrou uma vasculite granulomatosa necrotizante de células gigantes multinucleadas. A interpretação dos padrões histopatológicos mostrou uma evolução temporal gradativa de vasculite granulomatosa comprometendo predominantemente segmento cefálico, altamente favorável a granulomatose de Wegener.
Devido ao quadro clínico, ao resultado histopatológico e do c-ANCA, foi estabelecido, finalmente, o diagnóstico de granulomatose de Wegener, na sua forma limitada. A paciente iniciou tratamento com ciclosporina 100 mg/dia e prednisona 80 mg/dia, apresentando melhora sensível da proptose, após dois anos de acompanhamento.
Caso 2
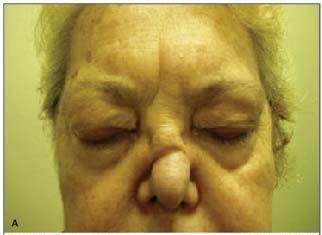
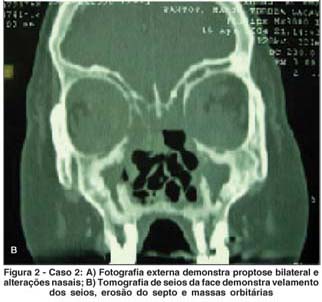
MTLS, sexo feminino, 60 anos, natural e procedente de São Paulo, aposentada. Queixa de proptose bilateral há 3 anos e sensação de corpo estranho nos olhos há 1 ano. História pregressa: Há três anos iniciou quadro de proptose bilateral (Figura 2A), sendo tratada com 60 mg/dia de prednisona, com melhora parcial no início do tratamento. Foi submetida ao uso de ciclofosfamida há 2 anos, com melhora mais evidente do quadro, porém com interrupção do tratamento há 1 ano por neutropenia. Passou a notar piora da proptose bilateralmente e a apresentar sensação de areia nos olhos. Antecedentes pessoais: sinusite de repetição há 15 anos, tendo sido submetida a cinco cirurgias entre 1992 e 1998. Há 6 anos é acompanhada com diagnóstico de pseudotumor orbitário, sendo tratada inicialmente com corticosteróides. Foi submetida à TC tórax que mostrou nódulo em lobo superior esquerdo, que foi biopsiado posteriormente, sendo compatível com vasculite granulomatosa necrotizante, ocasião onde foi feito o diagnóstico de GW. Há 2 anos iniciou tratamento com ciclofosfamida, porém desenvolveu intolerância e piora do quadro ocular, sendo então, encaminhada para a Oftalmologia. Antecedentes oftalmológicos: Proptose bilateral há 3 anos; Dacriocistite à esquerda há 2 anos. Exames prévios: TC seios da face: velamento dos seios, erosão septo, massas orbitárias (Figura 2B); TC tórax: nódulo em LSE, elevação da cúpula D. Exame oftalmológico: Proptose bilateral; presença de xantelasmas em pálpebras superiores; Motricidade ocular extrínseca: sem alterações; Reflexos fotomotores: sem alterações; Acuidade visual corrigida de 20/20 em ambos os olhos (refração estática OD -2,25 DE e OE -2,50 DE); Biomicroscopia: ceratite puntata difusa, com afilamento periférico superior e acúmulo de muco em ambos os olhos; PIO: 16/15 mmHg; FO: normal AO
Há 1 ano, vem fazendo uso de metotrexate e prednisona 80 mg/dia, além do uso tópico de colírio lubrificante e de acetilcisteína apresentando melhora da proptose e das queixas clínicas.
Caso 3
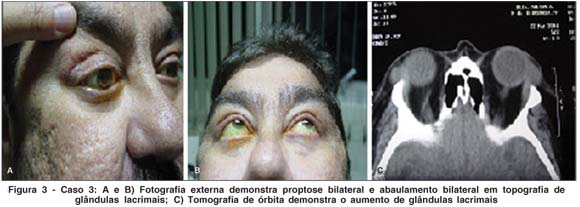
NR, sexo masculino, 46 anos, natural e procedente de São Paulo. Queixa de abaulamento em canto lateral de pálpebra superior bilateralmente há 1 ano. História pregressa: Há um ano iniciou quadro de abaulamento em pálpebras superiores bilateralmente (Figura 3A e 3B). Foi submetida à TC (Figura 3C) e à biópsia em topografia de glândula lacrimal, sendo compatível com GW. Antecedentes pessoais: Paciente acompanhado na Reumatologia com diagnóstico de GW, com acometimento renal, pulmonar e de seios da face há 4 anos. Antecedentes oftalmológicos: ndn. Exame oftalmológico: Proptose bilateral; edema e aumento de volume da glândula lacrimal bilateralmente; Motricidade ocular extrínseca: sem alterações; Reflexos fotomotores: sem alterações; Acuidade visual corrigida de 20/20 em ambos os olhos (refração estática OD -1,25 DE e OE -1,50 DE); Biomicroscopia: ndn; PIO: 16/15 mmHg; FO: normal AO.
Caso 4
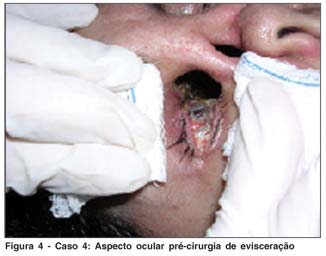
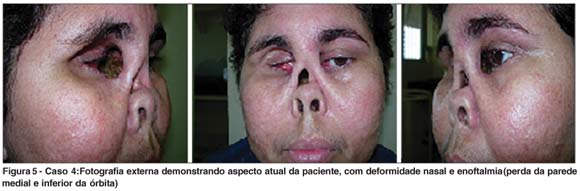
EAF, 32 anos, sexo feminino, natural e procedente de Parnaíba (PI). Queixa de diminuição da visão à direita + dor + fotofobia há 1 ano. História pregressa: Há 1 ano, apresentou diminuição da visão à direita, acompanhada de intensa dor, fotofobia e presença de secreção purulenta, sendo diagnosticada ceratite de exposição com conseqüente perfuração ocular, sendo submetida à transplante de córnea. Há 6 meses, houve piora do quadro ocular, com evolução para evisceração de olho direito (Figura 4). Atualmente está em tratamento com corticosteróide + pulsoterapia com imunoglobulina (Figura 5). Antecedentes pessoais: Há 4 anos início de episódios de repetição de rinorréia amarelada + cefaléia + dores nos ossos da face. Nesta ocasião foi levantada hipótese diagnóstica de GW, confirmada pelo c-ANCA + e TC tórax que mostrou um nódulo pulmonar. Iniciou tratamento com metotrexate + Meticorten® de forma irregular há 18 meses, houve piora do acometimento de vias aéreas superiores, com intensificação da deformidade nasal, evolução para enoftalmia (perda da parede medial e inferior da órbita), lagoftalmo e expressiva exposição ocular.
DISCUSSÃO
O primeiro caso de GW foi descrito por Heinz Klinger em 1931. Entre 1936 e 1939, Fried Wegener descreveu três outros casos, sendo reconhecida a desordem como uma forma de vasculite(8). Apresenta etiologia desconhecida, sendo considerada como uma reação de hipersensibilidade. Não há associação com doença alérgica, localização geográfica ou exposição ocupacional(8). Existe uma associação entre GW e HLA-B8 e HLA-DR2, indicando uma predisposição familiar(9).
Pode ocorrer de maneira sistêmica ou localizada e é caracterizada por vasculite necrosante das pequenas artérias e veias com formação de granulomas(1-2). Na sua forma típica, há dano sistêmico com envolvimento de trato respiratório superior e inferior, seguido por falência renal devido à glomerulonefrite necrotizante. Esta forma costuma ser fatal se não tratada(9). A forma atípica ou localizada costuma poupar os rins, apesar de estudos recentes demonstrarem que mesmo nestas formas existam lesões renais subclínicas(6).
A doença apresenta discreto predomínio no sexo masculino(10-11), sendo mais freqüente entre a quarta e quinta décadas(12), apesar de relatos de GW em crianças(13-14) e idosos(13,15) .
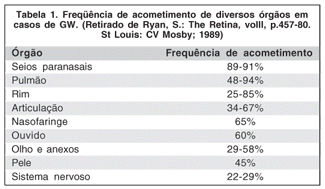
As manifestações sistêmicas da GW são muito variadas. Sinais e sintomas em trato respiratório são manifestações iniciais comuns da doença, e incluem obstrução nasal, epistaxe, sinusite, otite média e lesões em mucosa, que podem acabar em perfuração do septo nasal, levando à deformidade do nariz em forma de sela. Úlceras orais, gengivite e envolvimento de ossos alveolares é descrito(11). O seio maxilar é comumente acometido, sendo o espessamento de sua mucosa um dos sinais radiológicos mais precoces. Sintomas como tosse, dispnéia, dor torácica e hemoptise refletem o envolvimento pulmonar(10-11).
O comprometimento renal é tardio e freqüentemente causa morte em pacientes sem tratamento adequado(10,15). Isso explica a menor mortalidade nos casos atípicos (ou localizados). Paralisias de quinto e sétimo nervos são as manifestações neurológicas mais comuns(10-11). Cerca de 40 a 50% dos pacientes são portadores de lesões de pele equimóticas, ulcerativas ou na forma de nódulos subcutâneos(10-11). Aproximadamente dois terços dos pacientes se queixarão de artralgia ou apresentarão artrite(11).
A tabela 1 sumariza a freqüência de acometimento dos órgãos mais comumente envolvidos na GW.
O quadro ocular na GW pode ser também muito variado. O envolvimento orbitário da GW tem uma incidência que varia entre 40 e 64% dos casos e normalmente ocorre por extensão do acometimento em trato respiratório superior ou por vasculite focal envolvendo estruturas oculares(8). A apresentação orbitária pode ser idêntica a pseudotumor orbitário idiopático, exceto pelo fato que a apresentação bilateral ser mais comum na GW(15). O envolvimento da glândula lacrimal se manifesta como uma dacrioadenite, com boa resposta terapêutica aos corticosteróides, como no caso 3. Os casos descritos ilustram a dificuldade em se firmar o diagnóstico diferencial entre GW e pseudotumor.
O nervo óptico pode estar envolvido diretamente pelo processo vasculítico ou por etiologia compressiva. Retinite com hemorragias, edema, exsudatos algodonosos, e espessamento coroidal pode ser observada mesmo sem acometimento orbitário ou renal(10).
Como manifestações em segmento anterior, destaca-se conjuntivite, episclerite, esclerite anterior e posterior, e ceratite(7,10,16-17). Em formas severas, podem ser observadas complicações como esclerite necrosante ou ceratite ulcerativa periférica.
A biópsia representa papel de destaque no diagnóstico da GW. A vasculite geralmente envolve arteríolas e vênulas de pequeno calibre, podendo ser necrotizante ou granulomatosa, com infiltrado inflamatório misto e padrão geográfico de necrose tecidual(18). Muitas vezes ocorre dificuldade diagnóstica, mesmo em relação ao exame anátomo-patológico, sendo necessárias revisôes como nos casos 1 e 2.
A presença de anticorpos anti-citoplasma de neutrófilos (c-ANCA) é útil quando apresenta resultados positivos, auxiliando ao diagnóstico de formas limitadas atípicas (apresentando nestes casos 60 a 70% de positividade)(18). Enquanto o c-ANCA apresenta boa sensibilidade para GW, o p-ANCA está presente na poliarterite nodosa, síndrome de Churg-Strauss, e outras vasculites, auxiliando desta forma ao diagnóstico diferencial(18).
O envolvimento orbitário da GW deve ser distinguido de outras vasculites e doenças granulomatosas, além de doenças linfoproliferativas. Os principais diagnósticos diferenciais são: poliarterite nodosa, sarcoidose, granulomatose linfomatóide, e reticulose polimórfica. Quadros infecciosos como lues, tuberculose e fungo devem ser afastados antes da introdução da terapêutica apropriada, que visa a imunossupressão do paciente.
Antes da utilização de agentes imunossupressores, a GW era considerada patologia fatal. A média de sobrevivência era de 5 meses, e a mortalidade após 1 ano de 82%. Com a introdução de drogas citotóxicas, este panorama se alterou drasticamente. O tratamento é normalmente iniciado com prednisona 1 mg/kg peso, seguido pela introdução de ciclofosfamida 1 a 2 mg/kg peso. O corticóide é mantido até que haja controle da doença, e então retirado gradualmente. Tratamento com ciclofosfamida é mantido por 1 ano após completa remissão dos sintomas. Segundo o National Health Institute, este esquema possibilita a remissão da doença em 93% dos casos(6,17-18). Quando não há tolerância a ciclofosfamida, pode ser usado o metotrexate em ciclos semanais associado a corticosteróide(6).
A vasculite da região orbitária e a fibrose resultam em enoftalmia e exposição ocular de difícil tratamento, sendo que muitas vezes evoluem para região do bulbo, como ocorreu no caso 4, a despeito do tratamento sistêmico com imunossupressores.
Descrevem-se quatro casos de GW com apresentação atípica (ou localizada). O estudo anatomopatológico e a pesquisa do c-ANCA foram primordiais para o diagnóstico da doença.
Este trabalho mostra a dificuldade clínica e histológica no diagnóstico do comprometimento orbitário na GW.
REFERÊNCIAS
1. Goodman GC, Churg J. Wegener´s Granulomatosis: pathology and review of the literature. AMA Arch Pathol. 1954;58(6):533-53.
2. Fauci AS, Wolf SM. Wegener´s Granulomatosis: studies in eighteen patients and a review of the literature. Medicine (Baltimore). 1973;52(6):535-61.
3. Carrington CB, Liebow A. Limited forms of angiitis and granulomatosis of Wegener´s type. Am J Med. 1966;41(4):497-527.
4. Coppeto JR, Yamase H, Monteiro ML. Chronic ophthalmic Wegener´s granulomatosis. J Clin Neuroóphthalmol. 1985;5(1):17-25.
5. Bullen CL, Liesegang TJ, McDonald TJ, DeReeme RA. Ocular complications of Wegener´s granulomatosis. Ophthalmology. 1983;90(3):279-90.
6. Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener´s Granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983;98(1):76-85.
7. Haynes BF, Fishmann ML, Fauci AS, Wolff SM. The ocular manifestations of Wegener´s Granulomatosis. Fifteen years experience and review of the literature. Am J Med. 1977;63(1):131-41.
8. Lanza JT, Ku Y, Lucente FE, Har-El G. Wegener's granulomatosis of the orbit: lacrimal gland involvement as a major sign. Am J Otolaryngol. 1995;16(2):119-22.
9. Ataman M. Sarioglu T, Shahidi H, Gursel B. Wegener's granulomatosis: case report and review of the literature. Rhinology. 1994;32(2):92-7.
10. Fauci A, Haynes B, Costa J, Katz P, Wolff SM. Lymphomatoid granulomatosis: Prospective clinical and therapeutic experience over 10 years. N Engl J Méd. 1982;306(2):68-74.
11. Cupps T, Fauci A. Wegener's granulomatosis. In: Smith L, editor. Major Problems in Internal Medicine. Philadelphia: WB Saunders; 1981. v.21, p.72-87.
12. Spalton DJ, Graham EM, Page NG, Sanders MD. Ocular changes in limited forms of Wegener's granulomatosis. Br J Ophthalmol. 1981;65(8):553-63.
13. Jakobiec FA, Font RL. Orbit. In Spencer WH, editor. Ophthalmic pathology, 3rd ed. Philadelphia: WB Saunders; 1986. p.2459-60.
14. Hardwig PW, Bartley GB, Garrity JA. Surgical management of nasolacrimal duct obstruction in patients with Wegener's granulomatosis. Ophthalmology. 1992;99(1):133-9.
15. Soukiasian SH, Foster CS, Niles JL, Raizman MB. Diagnostic value of anti-neutrophil cytoplasmic antibodies in scleritis associated with Wegener's granulomatosis. Ophthalmology. 1983;99(1):125-32.
16. Fauci AS, Haynes BF, Katz P. The spectrum of vasculitis: clinical, pathologic, immunologic, and therapeutic considerations. Ann Intern Med. 1978;89(5 Pt 1):660-76.
17. Pulido JS, Goeken JA, Nerad JA, Sobol WM, Folberg R. Ocular manifestations of patients with circulating antineutrophil cytoplasmic antibodies. Arch Ophthalmol. 1990;108(6):845-50.
18. Specks U, Wheatley C, McDonald TJ, Rohrbach MS, DeRemee Ra. Anticytoplasmic autoantibodies in the diagnosis and follow-up of Wegener's granulomatosis. Mayo Clin Proc. 1989;64(1):28-36.
Endereço para correspondência:
Ivana Cardoso Pereira
Rua Oscar Freire, 1702 - Apto. 64
São Paulo (SP) CEP 05409-011
E-mail: [email protected]
Recebido para publicação em 03.07.2006
Aprovação em 24.08.2007
Última versão recebida em 23.10.2007
Trabalho realizado na Universidade de São Paulo - USP - São Paulo (SP) - Brasil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Ana Estela Besteti Pires Ponce Sant'Anna sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.