Arq. Bras. Oftalmol. 2007; 70 (3): 10.1590/S0004-27492007000300031
Total: 3441
Raul Nunes Galvarro Vianna1; Daniela Socci2; Márcio Bittar Nehemy3; Jean Deschênes4; Miguel Noel Burnier Jr5
DOI: 10.1590/S0004-27492007000300031
ABSTRACT
Several entities must be considered when a patient presents with a white dot syndrome. In most cases these can be distinguished from one another based on the appearance or distribution of the lesions, the clinical course, or patient variables such as age, sex, laterality, and functional and image examinations. In this paper we review the distinctive and shared features of the white dot syndromes, highlighting the clinical findings, diagnostic test results, proposed etiologies, treatment, and prognosis.
Keywords: Choroid diseases; Pigmentation disorders; Eletroretinography; Retinal diseases; Syndrome; Fluorescein angiography; Glucocorticoids; Macula lutea; Visual acuity
RESUMO
Várias doenças devem ser consideradas quando nos deparamos com paciente com uma entidade clínica incluída no grupo das "síndromes dos pontos brancos retinianos". O diagnóstico diferencial na maioria das vezes é baseado na aparência e/ou na distribuição das lesões, no curso clínico, ou por algumas variáveis relacionadas ao paciente, tais como idade, sexo, lateralidade, bem como por meio de exames funcionais e de imagem. O presente artigo revisa os achados clínicos das doenças que fazem parte do grupo das "síndromes dos pontos brancos retinianos", enfatizando as similaridades e as diferenças entre essas entidades. Os exames complementares, bem como a etiologia, o tratamento e o prognóstico de cada uma delas são descritos e comentados.
Descritores: Doenças da coróide; Doenças pigmentares; Eletrorretinografia; Doenças da retina; Síndrome; Angiografia fluoresceínica; Glicocorticóides; Glicocorticóides; Acuidade visual
ATUALIZAÇÃO CONTINUADA
Síndromes dos pontos brancos retinianos
Raul Nunes Galvarro ViannaI; Daniela SocciII; Márcio Bittar NehemyIII; Jean DeschênesIV; Miguel Noel Burnier JrV
IAssociate Professor, Head - Uveitis, Retina and Intra-ocular Tumors Unit, Department of Ophthalmology - Universidade Federal Fluminense - UFF - Niterói (RJ) - Brazil
IIStaff Physician, Uveitis Service - Universidade Federal do Rio de Janeiro - UFRJ - Rio de Janeiro (RJ) - Brazil
IIIAssociate Professor of Ophthalmology, Head Retina and Vitreous Unit, Department of Ophthalmology - Universidade Federal de Minas Gerais - UFMG - Belo Horizonte (MG) - Brazil
IVProfessor of Ophthalmology, Department of Ophthalmology - McGill University Health Center - Montreal - Quebec - Canada
VProfessor of Ophthalmology, Head-Department of Ophthalmology - McGill University Health Center - Montreal - Quebec - Canada
ABSTRACT
Several entities must be considered when a patient presents with a white dot syndrome. In most cases these can be distinguished from one another based on the appearance or distribution of the lesions, the clinical course, or patient variables such as age, sex, laterality, and functional and image examinations. In this paper we review the distinctive and shared features of the white dot syndromes, highlighting the clinical findings, diagnostic test results, proposed etiologies, treatment, and prognosis.
Keywords: Choroid diseases/diagnosis; Pigmentation disorders/diagnosis; Eletroretinography; Retinal diseases/diagnosis; Syndrome; Fluorescein angiography; Glucocorticoids/therapeutic use; Macula lutea/pathology; Visual acuity
RESUMO
Várias doenças devem ser consideradas quando nos deparamos com paciente com uma entidade clínica incluída no grupo das "síndromes dos pontos brancos retinianos". O diagnóstico diferencial na maioria das vezes é baseado na aparência e/ou na distribuição das lesões, no curso clínico, ou por algumas variáveis relacionadas ao paciente, tais como idade, sexo, lateralidade, bem como por meio de exames funcionais e de imagem. O presente artigo revisa os achados clínicos das doenças que fazem parte do grupo das "síndromes dos pontos brancos retinianos", enfatizando as similaridades e as diferenças entre essas entidades. Os exames complementares, bem como a etiologia, o tratamento e o prognóstico de cada uma delas são descritos e comentados.
Descritores: Doenças da coróide/diagnóstico; Doenças pigmentares/diagnóstico; Eletrorretinografia; Doenças da retina/diagnóstico; Síndrome; Angiografia fluoresceínica; Glicocorticóides/uso terapêutico; Macula lutea/patologia; Acuidade visual
INTRODUCTION
The so-called white dot syndromes (WDS) are a group of ocular disorders characterized by the presence of whitish lesions in the choroid, retinal pigment epithelium (RPE), and/or sensory retina. Despite the fact that many infectious and noninfectious inflammatory diseases may present with multifocal chorioretinal lesions, the entities included in the WDS share some features which make them a particular group of ocular disorders(1-2). In fact, the WDS would be better labeled as idiopathic inflammatory multifocal chorioretinopathies, since with the exception of diffuse unilateral subacute neuroretinitis, their causes are still unknown. The WDS group is composed of the following diseases: acute posterior multifocal placoid pigment epitheliopathy, serpiginous choroiditis, diffuse unilateral subacute neuroretinitis, birdshot chorioretinopathy, multiple evanescent white dot syndrome, punctate inner choroidopathy, multifocal choroiditis and panuveitis, and acute idiopathic exudative polymorphous vitelliform maculopathy.
The correct diagnosis of WDS is important because the management is totally different from one another. Some of them are self-limited and have good visual outcomes without treatment, while others are associated with serious retinal and choroidal sequelae, which can result in severe visual loss even after adequate immunosuppressive therapy.
The aim of this paper is to review the distinctive and shared features of the WDS, highlighting the clinical findings, diagnostic test results, proposed etiologies, treatment, and prognosis.
ACUTE POSTERIOR MULTIFOCAL PLACOID PIGMENT EPITHELIOPATHY (APMPPE)
APMPPE is the prototype of WDS and was first described by Gass in 1968(3). The condition typically affects young healthy adults, between 20-50 years of age. Some series show that women are more affected than men(4). Most patients have bilateral involvement, but unilateral cases have been reported. A history of a viral-like illness, two or three weeks before the onset of the ocular symptoms has been described, but it is not a rule. Mild anterior chamber cells are occasionally seen but vitreous cells are present in one third of affected eyes(4). The fundus picture is characterized by multiple, circumscribed, yellow-white lesions situated primarily in the posterior pole at the level of the RPE (Figure 1). Within a couple of weeks the acute lesions are replaced by varying degrees of RPE atrophy and hyperpigmentation.
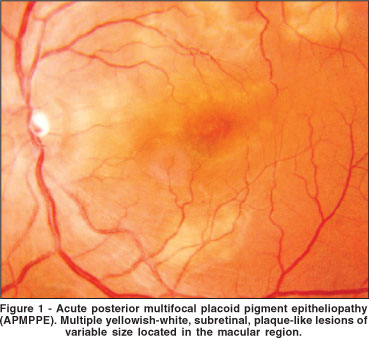
The diagnosis is based on the typical fundoscopic and fluorescein-angiographic findings. Fluorescein angiography shows hypofluorescence of the lesions in the early phase followed by late staining(3).
The hypofluorescence is probably related to both the gray-white opacification of the RPE and choroidal nonperfusion. In fact, it has been suggested that there is an obstruction at the level of the precapillary arterioles due to choroidal vasculitis(5). Interestingly, indocyanine green angiography (ICG) of patients with APMPPE reveals hypofluorescence of the active and healed lesions, highlighting the role of choroidal nonperfusion or infarction of the choroid(6).
The cause of APMPPE remains speculative. The relative frequency of an upper respiratory tract infection in the recent past points to a possible viral etiology. Nevertheless, APMPPE may be just an unspecific fundus picture, since it has been described in association with syphilis, sarcoidosis, tuberculosis, Crohn's disease, Wegener's granulomatosis, and other less frequent disorders(7-8). Therefore, in patients with an APMPPE-like fundus in Brazil, we do recommend systemic investigation to rule out infectious disorders. Special attention must be given to patients with APMPPE and severe headache or other neurologic sign or symptom, because these features may indicate cerebral vasculitis(9).
In general no treatment is necessary for APMPPE; the disease seems to be self-limited. Some ophthalmologists use systemic corticosteroids in cases in which the macula is affected, but no convincing evidence exists that corticosteroids speed visual recovery or improve visual outcome. Although many patients initially present with severe visual loss, a final visual acuity of 20/40 or better is the rule(4). Despite this good final visual acuity, an impressive number of patients continue to complain of scotomas, metamorphopsia, decreased vision, floaters and chronic redness of the eye(4). Recurrences of APMPPE are rarely seen. Choroidal neovascularization (CNV) may complicate the picture in some cases(10).
SERPIGINOUS CHOROIDITIS
Serpiginous choroiditis (SC) is a rare, usually bilateral chronically recurring inflammatory disease that affects the inner choroid and the RPE. SC usually begins in the peripapillary area and spreads centrifugally, in a snake-like manner, over a period of months or years (Figure 2)(10-11). In rare instances the lesions begin in the macular region, and this clinical presentation is associated with a worse visual prognosis. The presentation is in the fourth to sixth decades(10). Men and women are equally affected(10).
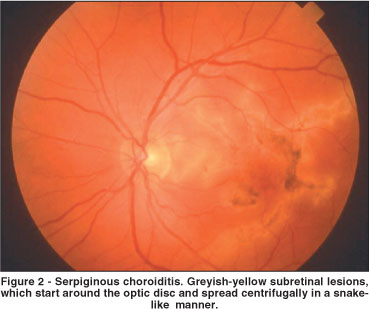
Ophthalmoscopically the active lesion appears as yellow-gray areas contiguous with, or satellites to, existing areas of chorioretinal atrophy. Fluorescein angiography shows early hypofluorescence and late hyperfluorescence of active lesions(10-11). ICG also shows hypofluorescence of the active lesions(12). It is possible that hypofluorescence of the lesions could result from a combination of choroidal nonperfusion and blockage by exudation or edema at the level of the outer retina, RPE and choriocapillaris. Interestingly, ICG angiography usually reveals a larger zone of involvement than can be seen on fluorescein angiography.
Although recent reports suggest that infectious agents may play a role in the pathogenesis of SC (e.g. herpes viruses, tuberculosis), its cause is still unknown(13-14). Nevertheless, the main controversy about SC is related to its treatment. The therapy with immunosuppressive agents is regarded as the best option to treat active SC. Indeed, current evidences suggest that immunologic mechanisms are involved in the pathogenesis of SC. Histopathologic studies have demonstrated diffuse and focal infiltrates of lymphocytes in the choroid, particularly at the margin of the serpiginous lesions, suggesting an inflammatory component of the disease(15). This is the rationale for the use of antinflammatory and immunosuppressive therapies for SC.
Some authors believe that systemic and periocular corticosteroids are enough for the treatment of SC(16). However, recent long-term follow-up studies have suggested that therapy with immunosuppressive agents is the best option to treat active SC, as steroids alone did not prevent recurrences(17-19). In SC it is important to prevent recurrences to preserve retinal function, especially if the lesion is near the macular region. Previous papers on the natural course of the disease have demonstrated that loss of vision is common because of repeated recurrences of choroidal inflammation(17-19). Therefore, the International Uveitis Study Group (IUSG) is recommending the use of immunosuppressive agents in patients with active SC(20). They believe that although this disorder may initially respond well to corticosteroids, the long-term prognosis and the morbidity associated with this disease are unacceptable. Unfortunately, no controlled trials comparing various treatments with these drugs are available to date. Hooper and Kaplan suggested the use of a combination of azathioprine (AZA), cyclosporine, and prednisone, the so-called "triple therapy", for the treatment of active SC(21). They have observed a rapid remission of active disease in five patients. However, recurrence of activity occurred in two patients after treatment was stopped but resolved after treatment was resumed. Araujo et al., and Christmas et al., also reported relative success of this "triple therapy" in some patients with active SC(17,19). In a recent paper, Vianna et al., suggested that AZA (125 mg/day), in combination with oral corticosteroids, was a safe and effective drug to treat active SC(22). Alkylating agents (chlorambucil and cyclophosphamide) have already been described to be efficient in the management of active SC(18).
However, because of the potentially severe systemic side effects (including malignancies), the treatment with these agents is recommended for those patients with acute bilateral foveal-threatening lesions. Besides the high rate of recurrences, another important problem related to SC is CNV. This devastating complication is observed in up to 35% of cases, and may be managed by laser photocoagulation(10). Indocyanine-mediated phototrombosis in combination with intravitreal corticosteroids may be an optional therapy to manage such cases(23). Many cases of SC are relentlessly progressive, and foveal destruction eventually occurs. It has been postulated, however that the rate of recurrence and other complications (e.g. CNV) may be decreased by long-term immunosuppressive therapy(24).
Central visual loss has been reported to be as high as 50% of involved eyes(19). However, most patients with SC maintain central function in at least one eye.
DIFFUSE UNILATERAL SUBACUTE NEURORETINITIS (DUSN)
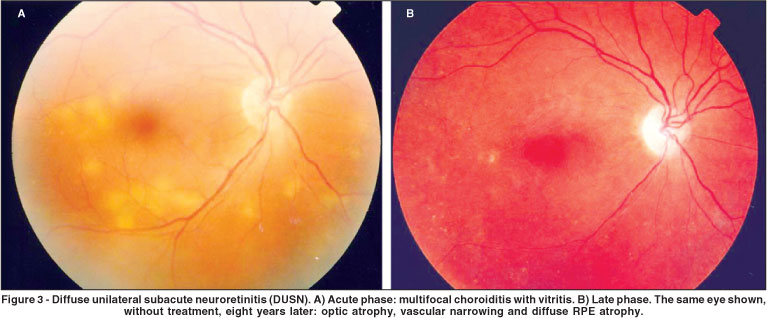
DUSN is an ocular inflammatory syndrome caused by a single subretinal nematode described initially by Gass et al. Occurring mainly in children and young adults, it affects one or, rarely, both eyes and usually results in severe loss of vision if left untreated(25). Unilateral visual loss, often with central or paracentral scotomata, is the primary symptom. Ophthalmoscopically, DUSN has two distinct stages(26). The early stage is characterized by inflammatory signs such as papillitis, multifocal chorioretinitis, and mild to moderate vitritis (Figure 3A). Fluorescein angiography demonstrates that the active lesions are hypofluorescent in the early frames and stain at a later time(10). Mild leakage may occur from the optic nerve. ICG in DUSN demonstrates hypofluorescence of the lesions in the early frames(27). However, unlike what is seen in fluorescein angiography, a few lesions remain hypofluorescent at the late times of the ICG examination. Electroretinography (ERG) amplitudes are moderately to severely reduced(10).
The late stage is characterized by narrowing vessels, optic nerve pallor, and focal or diffuse changes in the RPE (Figure 3B)(28). At this stage vision is usually hand movements.
The causative agent of DUSN is often suspected to be a toxocaral species, although ascarides from a variety of animal species can probably produce the disease(29-30). Studies by Souza et al., suggest that Ophidascaris, Polydelphis, Travassoascaris, and Hexametra must be considered as the etiological agent of DUSN in Brazil(32).
The diagnosis of DUSN in its early stage is important, because identification and destruction of the worm with laser photocoagulation prevents further visual loss and can occasionally lead to improved vision. Direct visualization of the nematode is generally made on clinical examination with fundus contact lenses. Another possibility is to use a scanning laser opthalmoscope, which provides a high contrast image that may facilitate visualization of the worm(31). Unfortunately, most patients in Brazil come to be examined in very late stages of the disease. In a series of 70 patients with DUSN from an endemic area of Northwestern Brazil, only four were diagnosed in the early stage of the disease(26). Besides the high degree of suspicion required to diagnose early-DUSN, other factors may delay treatment. For example, locating the worm in the retina can be a tedious and time-consuming task requiring many visits. Another complicating factor is that DUSN may mimic toxoplasmosis (outer punctate), tuberculosis or syphilis, diseases not uncommonly seen in South America. Souza et al., recently proposed an effective treatment using high doses of albendazol (400 mg/day-30 days) for the cases where the worm is not found(32). The treatment of late-stage DUSN is more complex. In fact, Garcia et al., reported on the relative inefficiency of laser treatment to improve vision of patients with late-stage DUSN(28). However, the authors emphasize that every patient with DUSN must be carefully evaluated in order to have the worm located and destroyed by laser, since this approach may avoid further visual loss.
BIRDSHOT CHORIORETINOPATHY
Birdshot chorioretinopathy (BC) is an uncommon form of chronic intraocular inflammation characterized by multiple, hypopigmented, postequatorial fundus inflammatory lesions(33). This disease was first reported by Ryan, Maumennee in 1980(33). According to these authors, the ocular fundus of affected patients resembled the scatter of a shotgun blast (birdshot). In fact, BC is not a new disorder since there are cases previously reported in the French literature, with the descriptive names of choriorétinopathie en taches de bougie (candlewax spots) and choriorétinopathie en grains de riz (rice grain pattern chorioretinopathy)(34-35). BC affects both eyes in virtually all cases and occurs in adults past the fourth decade of life, females more than males(33,36). Patients complain of blurred vision, floaters, central and peripheral photopsias, and later, nyctalopia and color blindness. The ocular fundus is characterized by scattered cream-colored or whitish hypopigmented lesions, cystoid macular edema, vasculitis, papillitis and chronic vitritis, but with minimal or no anterior segment involvement (Figure 4)(33)
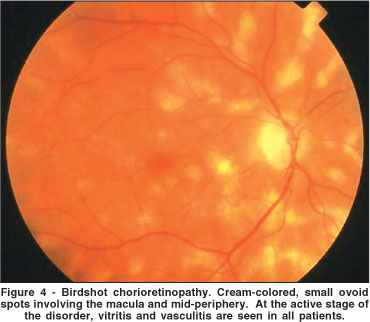
Fluorescein angiography reveals disc staining, vascular leakage, and cystoid macular edema(36). The hypopigmented patches usually do not show any significant change in normal background choroidal fluorescence. In the late phases of the angiogram, the hypopigmented lesions may appear mildly hyperfluorescent. In fact, these lesions are much more prominent ophthalmoscopically than angiographically. On the other hand, ICG shows the lesions very well as areas of blockage in the early to midphases of the angiogram, which may persist up to the late phases(37-38). The ERG reveals bilateral, moderately to severely depressed rod and cone function(39). Visual fields confirms the overall depression of retinal function(40). In fact, both tests (ERG and visual fields) are currently used in a systematic way (every six months) in order to detect disease reactivation(40). As the most common complication of BC is chronic macular edema, occurring in over 50% of the cases, optical coherence tomography (OCT) has become another important examination to evaluate the macular region of affected eyes(41).
The cause of BC is unknown, but virtually all patients with this disorder are HLA-A29 positive(42). This is the highest association of any HLA antigen with a human disease. The strong association with the HLA-A29, and the lymphocyte reactivity to retinal S-antigen suggests that BC may be an autoimmune disease.
The mainstay of treatment of BC patients has been the use of either periocular or systemic corticosteroids(36). However, the long-term use of systemic corticosteroids at the dose required to suppress the choroidal inflammation is likely to produce severe side effects, therefore, an immunosuppressive agent is warranted. Some recent papers suggest that immunosuppressive therapy would be the best option to manage BC patients(43-44). Uncontrolled case series have suggested that low-dose cyclosporine may be effective for BC. Vitale et al. found that the vitritis and the visual acuity were controlled in approximately 85% of the patients treated with cyclosporine(43). In contrast, in the same study, only 46% of the BC patients treated with corticosteroid therapy alone experienced an improvement in visual acuity during the follow-up period. However, due to cyclosporine's toxicity, other less toxic immunosuppressive therapy options were evaluated. Mycofenolate mofetil (MMF), an anti-metabolite agent, was used by Baltatzis et al., in patients with chronic ocular inflammatory disorders (including patients with BC). The inflammatory process was controlled in 75% of cases(44). In this study, the control of ocular inflammation with MMF as monotherapy was achieved in 35 (65%) patients, and a steroid-sparing effect was achieved in 29 (54%) patients. Additionally, Vianna et al., reported that MMF was effective in a BC patient refractory to AZA(45). Other treatment options are subtenon and/or intravitreal triamcinolone for refractory cystoid macular edema (CME)(41).
The prognosis is variable, but is better than in most other disorders with posterior uveitis. Visual loss may occur following chronic CME, CNV, vitreous hemorrhage, cataract and optic atrophy(36,46).
MULTIPLE EVANESCENT WHITE DOT SYNDROME (MEWDS)
MEWDS, initially described in 1984 by Jampol et al., is an acute, multifocal, usually unilateral retinopathy affecting mainly young women(47). A flu-like illness is present in about half of the cases.
Patients with MEWDS present with acute, unilateral, painless visual loss. The visual acuity ranges from 20/20 to 20/400 and most patients complain of the presence of a scotoma and associated photopsias, often in the temporal visual field. Ocular findings include a variable amount of vitritis, macular and/or optic disc edema, and, characteristically, several yellow-white dots at the level of the deep retina or RPE in the posterior pole (Figure 5). Besides the typical retinal lesions, a characteristic granular appearance of the fovea is present acutely, and the fovea usually does not return to a normal appearance(45).
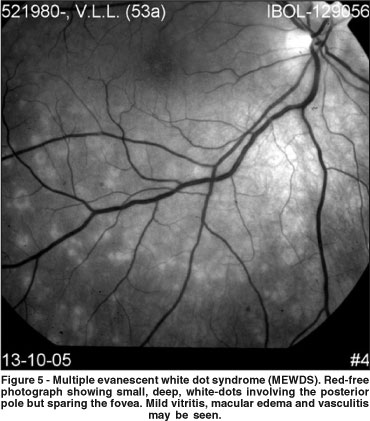
Fluorescein angiography reveals early and late hyperfluorescence of the white dots(47). Increased fluorescence of the optic disc may also be seen. Interestingly, OCT image in acute MEWDS may reveal macular edema, a sign just recently proposed as a possible cause of decreased vision in this disorder(48). ICG demonstrates multiple, round, hypofluorescent spots in the posterior pole(49). The number of spots observed on ICG may be more numerous than those seen ophthalmoscopically. The visual field typically reveals an enlarged blind spot and the ERG amplitudes are reduced(50). Overall, these features suggest that MEWDS affects the outer retina, the RPE and the choroid.
It has been suggested that the decreased vision observed in eyes with MEWDS may be related to transient metabolic disturbances at the level of the RPE - photoreceptor complex. Indeed, during the acute phase of the disease, the ERG a-wave and early receptor potential amplitudes are decreased in most affected patients, which suggests a primary involvement of the outer segments of photoreceptors(51). Multifocal electroretinogram shows areas of depression which correspond to scotomata while full field ERG shows a general depression(52). Keunen, Van Norren have used foveal densitometry and color matching to show that, even in those few patients with normal ERG findings, abnormalities exist during the active stage of MEWDS at the level of the cone photoreceptor outer segments(53). Nevertheless, focal ERG studies revealed delayed recovery of oscillatory potential, which also implies some inner retinal involvement(51). As previously mentioned in this paper, macular edema may also play a role in the transient decreased vision(48,54).
MEWDS has a self-limited course(47). Therefore, no specific treatment is necessary. The visual prognosis is excellent, although visual field loss, photopsia and metamorphopsia may take considerably longer to resolve (several months). Recurrences are rare and CNV may compromise the visual prognosis(55).
PUNCTATE INNER CHOROIDOPATHY (PIC)
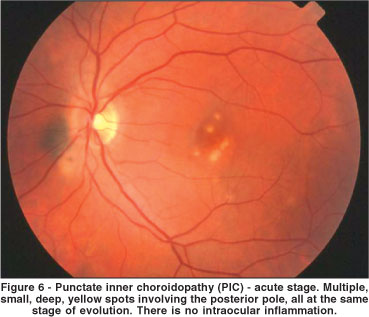
PIC, which was initially described by Watzke et al., in 1984, is a bilateral, ocular inflammatory disease that affects young healthy myopic women(56). Patients with PIC have acute symptoms of blurred vision, central or paracentral scotomata, photopsias, and sometimes peripheral visual fields loss. On ophthalmoscopic examination, small, yellow-white lesions (up to 300 µm in diameter) at the level of the choroid or RPE are present in the posterior pole (Figure 6)(56). Often a serous detachment of the retina overlies active lesions. Most of the lesions are located in the posterior pole or near periphery, but typically not in the far periphery. There are no cells or other signs of inflammation in the vitreous or anterior chamber(56). Patients with PIC usually have visual field defects such as central or paracentral scotoma, an enlarged blind spot or large areas of temporal visual field loss corresponding to numerous nasal chorioretinal scars. Fluorescein angiography of acute lesions reveals early hyperfluorescence and staining, and leakage into the subretinal space if an overlying serous detachment is present(56). ICG shows hypofluorescence of the lesions and may be helpful to differentiate acute lesions from incipient CNV(57). In fact, CNV emanating from scars, is observed in up to 25% of cases(58). Most of the CNV in PIC is extrafoveal. Systemic or periocular corticosteroids may be prescribed in cases of visual loss secondary to serous detachment, but usually the acute phase is self-limited and treatment is unnecessary. Laser photocoagulation of extrafoveal membranes is warranted to prevent visual loss, but long term follow-up of these treated eyes can reveal enlargement of the laser scar (most patients are myopic), which may compromise the visual prognosis(58). Photodynamic therapy, in combination or not with intravitreal corticosteroids, may be an option to treat selected cases of CNV in PIC(58). The visual prognosis of eyes with PIC that do not develop subfoveal CNV is good. Approximately 80% of eyes retain visual acuity of 20/25 or better.
MULTIFOCAL CHOROIDITIS AND PANUVEITIS (MCP)
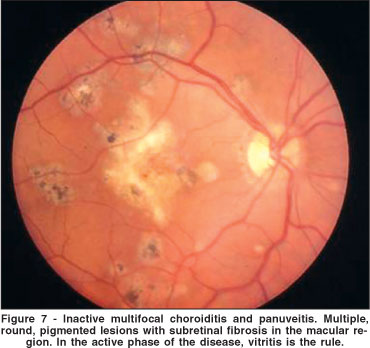
MCP, firstly described by Nozik, Dorsch in 1973, is an inflammatory disease of unknown cause characterized by fundus lesions similar to those seen in presumed ocular histoplasmosis syndrome, however, unlike the latter, vitreous and anterior chamber inflammation are present in affected eyes(59). MCP occurs predominantly in women between the second and sixth decades of life(60-61). The disease is bilateral in the majority of patients, although frequently it presents asymetrically, and many involved second eyes may be asymptomatic. Patients present with decreased visual acuity and/or photopsia. Most affected eyes have a variable amount of anterior segment inflammation and vitritis. This is an important feature to differentiate MCP from PIC. Ophthalmoscopically, choroidal lesions range in size up to 350 µm, arranged singly or in clumps located in the posterior pole and/or periphery (Figure 7). Acute lesions are yellow or gray and situated at the level of the RPE or deeper in the choroid. Older lesions are atrophic, "punched out", with variable amounts of pigment around or inside them(60-61).
The diagnosis is made by clinical examination. Fluorescein angiography reveals early blockage by acute, active, yellow lesions in the choroid, with late staining of these lesions(58). ICG shows hypofluorescent lesions which may cluster around the optic disc. Visual field testing may demonstrate scotomata that correspond to chorioretinal lesions or associated serous detachment of the retina and RPE(58). Large temporal field defects that do not correspond to visible fundus lesions are sometimes seen. Additionally, visual fields may reveal an enlarged blind spot as one of the initial manifestations of the disorder(58).
MCP tends to be a chronic disorder with recurrent bouts of inflammation. The treatment of MCP will depend on the degree of the intraocular inflammation. Topical corticosteroids and a mydriatic agent alone, or in combination with a posterior subtenon injection of corticosteroids may be an adequate therapy for mild to moderate cases(61). In more severe cases, oral prednisone and/or immunosuppressive agents are warranted. In fact, the wide spectrum of intraocular inflammation seen in eyes with MCP, suggests that the strategy for treatment must be modeled for each particular case. CME refractory to topical, periocular and systemic therapy may be managed with intravitreal corticosteroids. Photodynamic therapy has been recently used successfully to treat CNV associated with MCP(62).
It has been reported that the visual acuity of patients with MCP decreases with time(61). The visual loss can occur secondary to the inflammation itself (CME, CNV and cataract) and/or from corticosteroid-induced complications such as cataract and glaucoma. In some cases, subretinal fibrosis may develop and resemble subretinal fibrosis uveitis syndrome (SFS), a rare and visually devastating disorder(63). One can consider MCP and SFS as a unique disease but with a different intensity of subretinal fibrotic reaction.
Intriguingly, acute zonal outer occult retinopathy (AZOOR) has already been reported in association with MCP(64-65). Therefore, Gass has proposed that MCP (along with PIC, MEWDS, acute macular neuroretinopathy, acute annular outer retinopathy and acute idiopathic enlarged blind spot syndrome) be grouped within which he called the "AZOOR complex"(64). He has suggested that these diseases represent part of a spectrum of what is probably a single disease. In fact, AZOOR is seen in young females who complain of photopsia and present with peripheral loss of visual field, often in abrupt episodes. The disease may be bilateral or unilateral and may have a recurrent course. The fundus is initially normal, although areas of peripheral and peripapillary retina may develop mottled pigmentation, vascular narrowing and sheating. Nevertheless, these patients do not show neither white dots and/or plaques in the active phase of the disease nor chorioretinal scars in later stages. Affected eyes never get CNV. Patients with AZOOR have not been shown to respond to corticosteroids. For these reasons we agree with Jampol's opinion that each disease of the so-called AZOOR complex is a separable entity with different prognosis and different therapeutic regimens(66). In fact, the rare occurrence of two or more of these diseases in the same patient at different times (for example MEWDS and PIC) may be at least partially explained by the common genetic hypothesis of autoimmune/inflammatory disease proposed by Becker in 2001(67). This hypothesis states that patients with one of the AZOOR complex disorders share common nondisease specific gene clusters at specific genetic loci that predispose the patient to immune dysregulation and autoimmune disease(68). The interplay between immune dysregulation, specific environmental triggers and major histocompatibility antigens, explain some of the variability of the clinical course.
ACUTE IDIOPATHIC EXUDATIVE POLYMORPHOUS VITELLIFORM MACULOPATHY (AIEPVM)
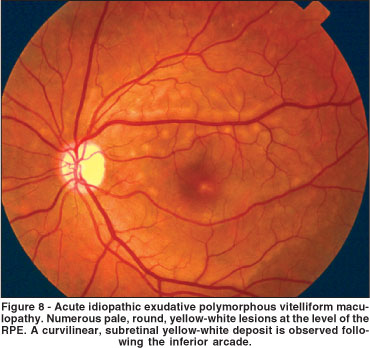
AIEPVM is a rare disorder initially described by Gass et al., in 1988(69). Typically, patients report intense headache and bilateral moderate vision loss. Ophthalmoscopy reveals multiple, round, yellow-white subretinal lesions at the level of the RPE, associated or not with serous detachment (Figure 8). In the acute phase of the disease, the round lesions show hyperfluorescence during the early phases of the fluorescein angiogram and late staining. Interestingly, the same pattern of fluorescence occurs in ICG(70). In both fluorescein angiography and ICG the hyperfluorescent round lesions correspond to the ophthalmoscopically seen lesions. Optical coherence tomography reveals anterior displacement of the photoreceptor layer by a hypereflective subretinal layer overlying a hyporeflective space above the RPE-choriocapillaris complex under all lesions and no subretinal fluid(71). The ERG amplitudes are usually normal, but the electrooculogram (EOG) may have a decrease of the Arden ratio(69-70). The above mentioned features indicate that the pathologic process is primarily located at the level of the RPE and choriocapillaris.
The cause and pathophysiology of AIEPVM still remain obscure. The best management of this disorder has been not established yet. Gass et al., treated two AIEPVM patients with systemic corticosteroids who later developed polymorphous, yellow subretinal deposits(69). On the other hand, Vianna et al. reported one patient who recovered full vision without treatment(70). These authors speculate that corticosteroids may play a role in the natural course of AIEPVM, worsening the ocular picture.
In fact, little is known about this entity, but the ocular features and the benign, self-limited clinical course suggests that AIEPVM is an acquired inflammatory disorder. For these reasons, AIEPVM has been recently suggested to be included in the WDS group(70).
REFERENCES
01. Vianna RNG, Muralha A, Muralha L, Moura LR, Burnier Jr MN. Flecked retina in systemic large-cell non-Hodgkin's lymphoma. Arq Bras Oftalmol. In press.
02. Muralha A, Muralha L, Curi AL, Muranaka E, Vianna RN. Coroidite criptocócica em síndrome de imunodeficiência adquirida. Rev Bras Oftalmol. 1998; 57(12):941-5.
03. Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80(2):177-85.
04. Vianna R, Van Egmond J, Priem H, Kestelyn P. Natural history and visual outcome in patients with APMPPE. Bull Soc Belge Ophtalmol. 1993;248 (1):73-6.
05. Deutman AF, Lion F. Choriocapillaris nonperfusion in acute multifocal placoid pigment epitheliopathy. Am J Ophthalmol. 1977;84(5):652-7.
06. Dhaliwal RS, Maguire AM, Flower RW, Arribas NP. Acute posterior multifocal placoid pigment epitheliopathy. An indocyanine green angiographic study. Retina. 1993;13(4):317-25.
07. Vianna RN, Ozdal PC, Deschenes J. Multifocal choroiditis-an unusual finding in Crohn's disease. Eur J Ophthalmol. 2004;14(4):345-9.
08. Quillen DA, Davis JB, Gottlieb JL, Blodi BA, Callanan DG, Chang TS, et al. The white dot syndromes. Am J Ophthalmol. 2004;137(3):538-50. Comment in: Am J Ophthalmol. 2004;138(4):686; author reply 686-7.
09. Wilson CA, Choromokos EA, Sheppard R. Acute posterior multifocal placoid pigment epitheliopathy and cerebral vasculitis. Arch Ophthalmol. 1988; 106(6):796-800.
10. Gass JDM. Stereoscopic atlas of macular diseases: diagnosis and treatment. 4th ed. St Louis: Mosby; 1997.
11. Laatikainen L, Erkkila H. Serpiginous choroiditis. Br J Ophthalmol. 1974; 58(9):777-83.
12. Giovannini A, Mariotti C, Ripa E, Scassellati-Sforzolini B. Indocyanine green angiographic findings in serpiginous choroidopathy. Br J Ophthalmol. 1996;80(6):536-40.
13. Priya K, Madhavan HN, Reiser BJ, Biswas J, Saptagirish R, Narayana KM, et al. Association of herpesviruses in the aqueous humor of patients with serpiginous choroiditis: a polymerase chain reaction-based study. Ocul Immunol Inflamm. 2002;10(4):253-61. Comment in: Ocul Immunol Inflamm. 2002;10(4):235-8.
14. Gupta V, Gupta A, Arora S, Bamberry P, Dogra MR, Agarwal A. Presumed tubercular serpiginouslike choroiditis; clinical presentations and management. Ophthalmology. 2003;110(9):1744-9.
15. Wu JS, Lewis H, Fine SL, Grover DA, Green WR. Clinicopathologic findings in a patient with serpiginous choroiditis and treated choroidal neovascularization. Retina. 1989;9(4):292-301.
16. Hoyng C, Tilanus M, Deutman A. Atypical central lesions in serpiginous choroiditis treated with oral prednisone. Graefes Arch Clin Exp Ophthalmol. 1998;236(2):154-6.
17. Araujo AA, Wells AP, Dick AD, Forrester JV. Early treatment with cyclosporin in serpiginous choroidopathy maintains remission and good visual outcome. Br J Ophthalmol. 2000;84(9):979-82.
18. Akpek EK, Jabs DA, Tessler HH, Joondeph BC, Foster CS. Successful treatment of serpiginous choroiditis with alkylating agents. Ophthalmology. 2002;109(8):1506-13.
19. Christmas NJ, Oh KT, Oh DM, Folk JC. Long-term follow-up of patients with serpinginous choroiditis. Retina. 2002;22(5):550-6.
20. Rao N. Management of intraocular inflammation. In: Ryan SJ, Schachat AP, editors. Retina. St Louis: Mosby; 2001. p.1032-8.
21. Hooper PL, Kaplan HJ. Triple agent immunosuppression in serpiginous choroiditis. Ophthalmology. 1991;98(6):944-51; discussion 951-2.
22. Vianna RN, Ozdal PC, Deschenes J, Burnier MN Jr. Combination of azathioprine and corticosteroids in the treatment of serpiginous choroiditis. Can J Ophthalmol. 2006;41(2):183-9.
23. Navajas EV, Costa RA, Farah ME, Cardillo JA, Bonomo PP. Indocyanine green-mediated photothrombosis combined with intravitreal triamcinolone for the treatment of choroidal neovascularization in serpiginous choroiditis. Eye. 2003;17(5):563-6.
24. Dees C, Arnold JJ, Forrester JV, Dick AD. Immunosuppressive treatment of choroidal neovascularization associated with endogenous posterior uveitis. Arch Ophthalmol. 1998;116(11):1456-61.
25. Gass JDM, Gilbert WR Jr, Guerry RK, Scelfo R. Diffuse unilateral subacute neuroretinitis. Ophthalmology. 1978;85(5):521-45.
26. Garcia CA, Gomes AH, Garcia Filho CA, Vianna RN. Early-stage diffuse unilateral subacute neuroretinitis: improvement of vision after photocoagulation of the worm. Eye. 2004;18(6):624-7.
27. Vianna RN, Onofre G, Ecard V, Muralha L, Muralha A, Garcia CA. Indocyanine green angiography in diffuse unilateral subacute neuroretinitis. Eye. 2006;20(9):1113-6.
28. Garcia CA, Gomes AH, Vianna RN, Souza Filho JP, Garcia Filho CA, Orefice F. Late-stage diffuse unilateral subacute neuroretinitis: photocoagulation of the worm does not improve the visual acuity of affected patients. Int Ophthalmol. 2005;26(1-2):39-42.
29. Goldberg MA, Kazacos KR, Boyce WM, Ai E, Katz B. Diffuse unilateral subacute neuroretinitis. Morphometric, serologic, and epidemiologic support for Baylisascaris as a causative agent. Ophthalmology. 1993;100(11):1695-701. Comment in: Ophthalmology. 1994;101(6):971-2.
30. Mets MB, Noble AG, Basti S, Gavin P, Davis AT, Shulman ST, et al. Eye findings of diffuse unilateral subacute neuroretinitis and multiple choroidal infiltrates associated with neural larva migrans due to Bbaylisascaris procyonis. Am J Ophthalmol. 2003;135(6):888-90.
31. Moraes LR, Cialdini AP, Avila MP, Elsner AE. Identifying live nematodes in diffuse unilateral subacute neuroretinitis by using the scanning laser ophthalmoscope. Arch Ophthalmol. 2002;120(2):135-8.
32. Souza EC, Casella AM, Nakashima Y, Monteiro ML. Clinical features and outcomes of patients with diffuse unilateral subacute neuroretinitis treated with oral albendazole. Am J Ophthalmol. 2005;140(3):437-445. Erratum in: Am J Ophthalmol. 2006;141(4):795-6. Erratum in: Am J Ophthalmol. 2006;141(4): 795-6.
33. Ryan SJ, Maumenee AE. Birdshot retinochoroidopathy. Am J Ophthalmol. 1980;89(1):31-45.
34. Franceschetti A, Babel J. La chorio-rétinite en "tache de bougie", manifestation de la maladie de Besnier-Boeck. Ophthalmologica. 1949;118(4-5):701-10.
35. Amalric P, Cuq G. [A very unusual form of chorioretinopathy, like grains of rice] Bull Soc Ophtalmol Fr. 1981 Jan;81(1):131-4. French.
36. Priem HA, Oosterhuis JA. Birdshot chorioretinopathy: clinical characteristics and evolution. Br J Ophthalmol. 1988;72(9):646-9.
37. Fardeau C, Herbort CP, Kullmann N, Quentel G, LeHoang P. Indocyanine green angiography in birdshot chorioretinopathy. Ophthalmology. 1999;106 (10):1928-34.
38. Vianna RN, Socci D, Deschênes J, Burnier MN Jr. The role of indocyanine green angiography in the diagnosis of birdshot chorioretinopathy. Rev Bras Oftalmol. 2006;65(4):242-45.
39. Priem HA, De Rouck A, De Laey JJ, Bird AC. Electrophysiologic studies in birdshot chorioretinopathy. Am J Ophthalmol. 1988;106(4):430-6.
40. Oh KT, Christmas NJ, Folk JC. Birdshot retinochoroiditis: long term follow-up of a chronically progressive disease. Am J Ophthalmol. 2002;133(5):622-9.
41. Socci D, Vianna RNG, Moura Brasil O. Optic coherence tomography in birdshot chorioretinopathy [poster]. In: World Ophthalmology Congress. São Paulo, 19-24 Feb. 2006.
42. Priem HA, Kijlstra A, Noens L, Baarsma GS, De Laey JJ, Oosterhuis JA. HLA typing in birdshot chorioretinopathy. Am J Ophthalmol. 1988;105(2):182-5.
43. Vitale AT, Rodriguez A, Foster CS. Low-dose cyclosporine therapy in the treatment of birdshot retinochoroidopathy. Ophthalmology. 1994;101(5):822-31.
44. Baltatzis S, Tufail F, Yu EN, Vredeveld CM, Foster CS. Mycophenolate mofetil as an immunomodulatory agent in the treatment of chronic ocular inflammatory disorders. Ophthalmology. 2003;110(5):1061-5.
45. Vianna RN, Al-Kharusi N, Deschenes J. The successful use of mycophenolate mofetil in a patient with active birdshot chorioretinopathy refractory to azathioprine therapy: case report. Arq Bras Oftalmol. 2004;67(6):957-9.
46. Vianna RN, Serop S, Priem HA. Atypical birdshot chorioretinopathy. Rev Bras Oftalmol. 1994;53(5):77-81.
47. Jampol LM, Sieving PA, Pugh D, Fishman GA, Gilbert H. Multiple evanescent white dot syndrome. I. Clinical findings. Arch Ophthalmol. 1984;102 (5):671-4.
48. Vianna RN, Soares Maia A, Rielo de Moura L. Optic coherence tomographic findings in multiple evanescent white dot syndrome. Submitted.
49. Obana A, Kusumi M, Miki T. Indocyanine green angiographic aspects of multiple evanescent white dot syndrome. Retina. 1996;16(2):97-104.
50. Sieving PA, Fishman GA, Jampol LM, Pugh D. Multiple evanescent white dot syndrome. II. Electrophysiology of the photoreceptors during retinal pigment epithelial disease. Arch Ophthalmol. 1984;102(5):675-9.
51. Horiguchi M, Miyake Y, Nakamura M, Fujii Y. Focal electroretinogram and visual field defect in multiple evanescent white dot syndrome. Br J Ophthalmol. 1993;77(7):452-5.
52. Chen D, Martidis A, Baumal CR. Transient multifocal electroretinogram dysfunction in multiple evanescent white dot syndrome. Ophthalmic Surg Lasers. 2002;33(3):246-9.
53. Keunen JE, Van Norren D. Foveal densitometry in the multiple evanescent white dot syndrome. Am J Ophthalmol. 1988;105(5):561-2.
54. Khurana RN, Albini T, Dea MK, Rao NA, Lim JI. Atypical presentation of multiple evanescent white dot syndrome involving granular lesions of varying size. Am J Ophthalmol. 2005;139(5):935-7.
55. Oh KT, Christmas NJ, Russel SR. Late recurrence and choroidal neovascularization in multiple evanescent white dot syndrome. Retina. 2001;21(2):182-4.
56. Watzke RC, Packer AJ, Folk JC, Benson WE, Burgess D, Ober RR. Punctate inner choroidopathy. Am J Ophthalmol. 1984;98(5):572-84.
57. Tiffin PA, Maini R, Roxburgh ST, Ellingford A. Indocyanine green angiography in a case of punctate inner choroidopathy. Br J Ophthalmol. 1996;80(1):90-1.
58. Folk JC, Walker JD. Multifocal choroiditis with panuveitis, diffuse subretinal fibrosis, and punctate inner choroidopathy. In: Ryan SJ, Schachat AP, Hinton DR, Wilkinson P, editors. Retina. 4th ed. St Louis: Mosby; 2006.
59. Nozik RA, Dorsch W. A new chorioretinopathy associated with anterior uveitis. Am J Ophthalmol. 1973;76(5):758-62.
60. Brown J Jr, Folk JC, Reddy CH, Kimura AE. Visual prognosis of multifocal choroiditis and panuveitis, punctate inner choroidopathy, and the diffuse subretinal fibrosis sndrome. Ophthalmology. 1996;103(7):1100-5.
61. Vianna RN, Ozdal PC, Souza Filho JP, Ventura MP, Saraiva VS, Deschenes J. Longterm follow-up of patients with multifocal choroiditis and panuveitis. Acta Ophthalmol Scand. 2004;82(6):748-53.
62. Spaide RF, Freund KB, Slakter J, Sorenson J, Yannuzzi LA, Fisher Y. Treatment of subfoveal choroidal neovascularization associated with multifocal choroiditis and panuveitis with photodynamic therapy. Retina. 2002;22(5): 545-9. Comment in: Retina. 2003;23(3):428; discussion 428.
63. Cantrill HL, Folk JC. Multifocal choroiditis associated with progressive subretinal fibrosis. Am J Ophthalmo. 1986;101(2):170-80.
64. Gass JD. Acute zonal occult outer retinopathy. Donders Lecture: The Netherlands Ophthalmological Society, Maastricht, Holland, June 19,1992. J Clin Neuroophthalmol. 1993;13(2):79-97.
65. Gass JD. Are acute zonal occult outer retinopathy and the white spot syndromes (AZOOR complex) specific autoimmune diseases? Am J Ophthalmol. 2003;135 (3):380-1. Comment on: Am J Ophthalmol. 2003;135(3):376-9.
66. Jampol LM, Wiredu A. MEWDS, MFC, PIC, AMN, AIBSE, and AZOOR: one disease or many? Retina. 1995;15(5):373-8.
67. Becker KG. The common genetic hypothesis of autoimmune inflammatory disease. Curr Opin Allergy Clin Immunol. 2001;1(5):399-405.
68. Jampol LM, Becker KG. White spot syndromes of the retina: a hypothesis based on the common genetic hypothesis of autoimmune/inflammatory disease. Am J Ophthalmol. 2003;135(3):376-9. Comment in: Am J Ophthalmol. 2003; 135(3):380-1.
69. Gass JD, Chuang EL, Granek H. Acute exudative polymorphous vitelliform maculopathy. Trans Am Ophthalmol Soc. 1988;86:354-66.
70. Vianna RN, Muralha A, Muralha L. Indocyanine green angiography in acute idiopathic exudative polymorphous vitelliform maculopathy. Retina. 2003; 23 (4):539-41.
71. Cruz-Villegas V, Villate N, Knighton RW, Rubsamen P, Davis JL. Optical coherence tomographic findings in acute exudative polymorphous vitelliform maculopathy. Am J Ophthalmol. 2003;136(4):760-3.
 Correspondence to:
Correspondence to:
Raul Nunes Galvarro Vianna
Rua Presidente João Pessôa, 346/201
Niterói (RJ) - Brasil CEP 24220-331
E-mail:[email protected]
Recebido para publicação em 26.08.2006
Aprovação em 26.02.2007
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência do Dr. João J. Nassaralla Jr. sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.
Trabalho realizado na Universidade Federal Fluminense - UFF - Niterói (RJ) - Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}