Arq. Bras. Oftalmol. 2007;70 (5 )
:851-853
| DOI: 10.1590/S0004-27492007000500022
Abstract
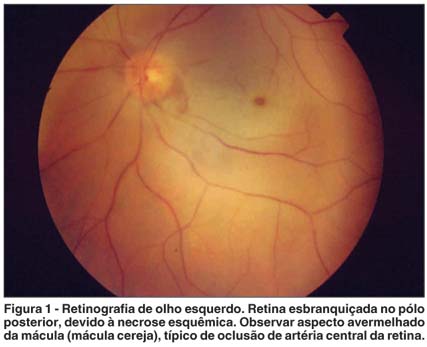
Os autores relatam o caso de um paciente com insuficiência coronariana que desenvolveu quadro de oclusão de artéria central da retina após ser submetido a cateterização cardíaca por via braquial e realização de cineangiocoronariografia. Este procedimento pode desencadear fenômenos embólicos oculares consistentes com o quadro descrito.
Keywords: Cateterismo cardíaco; Embolia; Oclusão da artéria retiniana; Cineangiografia; Fatores de risco; Relatos de casos
Arq. Bras. Oftalmol. 2007;70 (5 )
:871-874
| DOI: 10.1590/S0004-27492007000500027
Abstract
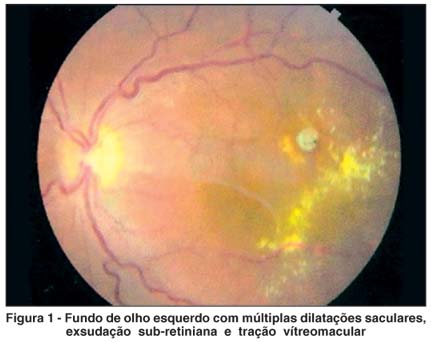
Telangiectasias retinianas são anormalidades vasculares primárias e idiopáticas caracterizadas por dilatações irregulares e incompetência dos vasos retinianos com variados graus de exsudação intra e sub-retiniana. O objetivo desse relato é documentar uma rara associação entre aneurisma miliar de Leber e síndrome de tração vítreomacular bem caracterizada à angiofluoresceinografia e tomografia de coerência óptica. O tratamento realizado foi fotocoagulação com laser de argônio nos aneurismas perimaculares e cirurgia de vitrectomia posterior via pars plana, o que resultou em melhora consistente da acuidade visual. O caso relatado confirma a importância da tomografia de coerência óptica em estudar a interface vítreorretiniana e suas alterações, o que permitiu abordagem completa da doença em questão.
Keywords: Telangiectasia; Aneurisma; Vasos retinianos; Angiofluoresceinografia; Fotocoagulação; Corpo vítreo; Tomografia de coerência óptica; Adulto; Relatos de casos
Arq. Bras. Oftalmol. 2007;70 (6 )
:1029-1033
| DOI: 10.1590/S0004-27492007000600028
Abstract
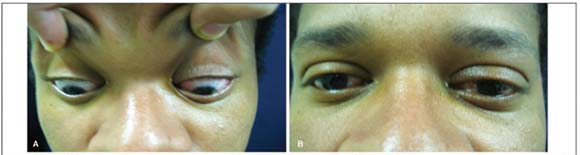
Inflamação orbital não-específica apresenta diversas formas clínicas. O envolvimento do segmento posterior do olho, geralmente, por contigüidade pode trazer sérios danos à função visual. A esclerite posterior, em geral, acarreta prejuízo permanente da visão e raramente evolui com glaucoma agudo. RELATO DO CASO: E.N., 24 anos, masculino, negro apresentando queixa de dor em OE há dez dias, acompanhada de diminuição da acuidade visual, mal-estar geral, náuseas e vômitos. Ao exame oftalmológico apresentava proptose, restrição da movimentação e edema na pálpebra superior de OE. AV c/c: 20/20 e CD 1,5m. À biomicroscopia, apresentava em OE hiperemia conjuntival, córnea com precipitados endoteliais, câmara anterior rasa, células e " flare" na câmara anterior 2+. Pressão intra-ocular (Po) de 14 mmHg em OD e 34 mmHg em OE. A gonioscopia em OE evidenciava ângulo fechado 360º, não se visualizando linha de Schwalbe. O mapeamento de retina revelava aumento da tortuosidade vascular e edema do pólo posterior. O tratamento para o glaucoma agudo foi instituído, ainda em ambiente hospitalar, e solicitados exames complementares. O exame de ultra-som ocular e TC de órbita revelaram espessamento difuso da parede ocular e da musculatura extrínseca ocular. Os demais exames apresentaram-se dentro da normalidade. A hipótese diagnóstica foi de inflamação orbitária anterior não-específica aguda com envolvimento do segmento posterior do globo ocular, complicado por glaucoma agudo. Instituiu-se tratamento com prednisona 60 mg/dia via oral. Após duas semanas do início da corticoterapia sistêmica, apresentava-se assintomático com nítida regressão da proptose, do quadro de esclerite e normalização da Po (11mmHg em AO). O presente caso, apesar de pouco freqüente, mostra que o glaucoma agudo pode estar presente em um quadro inflamatório orbitário e deve ser tratado com corticoterapia sistêmica, além da medicação tópica.
Keywords: Doenças orbitárias; Esclerite; Glaucoma de ângulo fechado; Relatos de casos
Arq. Bras. Oftalmol. 2011;74 (4 )
:255-257
| DOI: 10.1590/S0004-27492011000400005
Abstract
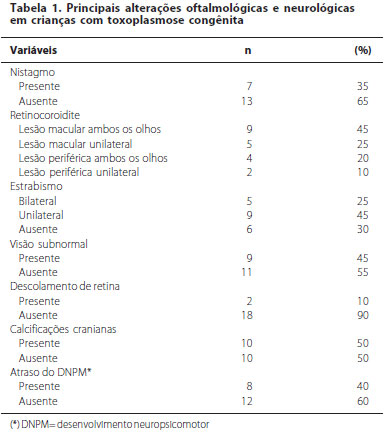
OBJETIVO: Conhecer as lesões oculares mais frequentes encontradas em crianças expostas à toxoplasmose congênita. MÉTODOS: Trata-se de um estudo retrospectivo, a partir de uma coorte histórica, de abordagem quantitativa. Foram avaliadas crianças encaminhadas de um serviço de infectologia pediátrica e inseridas apenas aquelas com diagnóstico confirmado de toxoplasmose congênita. A avaliação oftalmológica incluiu o mapeamento de retina sequencial, sob dilatação pupilar. RESULTADOS: Das 58 crianças presumivelmente expostas ao risco de doença durante a gestação, 20 apresentaram lesões oftalmológicas ao longo do primeiro ano de vida (34 olhos). Destas, 12 estavam assintomáticas ao nascimento. Estrabismo foi registrado em 14 crianças (70%). Em uma criança observou-se ptose palpebral e em outra diminuição da fenda palpebral (microftalmia). Retinocoroidite foi a complicação mais frequente, presente em todas as 20 crianças. Sete crianças apresentaram alterações unilaterais (35%) e 13 crianças apresentaram alterações bilaterais (65%), prevalecendo a localização no polo posterior e mácula. CONCLUSÃO: Retinocoroidite e estrabismo destacaram-se como importantes sequelas da toxoplasmose congênita.
Keywords: Toxoplasmose congênita; Coriorretinite; Criança; Infecções oculares parasitárias; Sinais e sintomas
Arq. Bras. Oftalmol. 2012;75 (5 )
:352-355
| DOI: 10.1590/S0004-27492012000500012
Abstract
OBJETIVO: Descrever as características clínicas e imaginológicas de duas famílias com a síndrome de Waardenburg, sendo uma do tipo I e outra do tipo II, enfatizando as manifestações oftalmológicas, bem como o padrão de herança genética. MÉTODO: Realizou-se um estudo clínico envolvendo as duas famílias afetadas pela síndrome de Waardenburg, sendo, através dos heredogramas, determinado o padrão de herança genética presente. Também foram realizadas análises oftalmológicas abordando a medida da acuidade visual, a presença de distopia cantorum (telecanto), a avaliação da coloração da íris e o mapeamento de retina, além de exames otológicos e dermatológicos. RESULTADOS: O heredograma da família afetada pela síndrome de Waardenburg tipo I revelou um modo autossômico dominante de transmissão. A condição estava presente em 85,71% dos pacientes. A distopia cantorum foi a alteração mais frequente, seguida pela mecha branca na pele da fronte, hipopigmentação da íris e da retina e surdez neurossensorial. A família com síndrome de Waardenburg tipo II apresentou 33,33% dos familiares com a alteração. Nenhum membro apresentou distopia cantorum e hipopigmentação de íris. Três pacientes apresentaram surdez neurossensorial (12,5%), associada ao topete branco e manchas acrômicas confluentes pelo corpo. CONCLUSÃO: O presente estudo mostra a importância do oftalmologista no auxílio do diagnóstico desta rara condição genética, uma vez que inclui alterações oftalmológicas como telecanto, hipopigmentação da íris e retina. A distopia cantorum é o principal critério diagnóstico para diferenciar o tipo I do II e deve ser feita por oftalmologista treinado. As famílias encontram-se em acompanhamento multiprofissional, tendo recebido orientações genéticas e os cuidados referentes à proteção ocular.
Keywords: Síndrome de Waardenburg; Doenças da íris; Doenças retinianas; Aconselhamento genético; Surdez; Pálpebras; Nariz; Relatos de casos
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.