Arq. Bras. Oftalmol. 2020;83 (5 )
:410-416
| DOI: 10.5935/0004-2749.20200080
Abstract
Objetivo: Avaliar as espessuras internas da retina e da coroide em pacientes com retinite pigmentosa precoce.
Métodos: Foram analisadas imagens de tomografia de coerência óptica de domínio espectral de 35 pacientes com retinite pigmentosa e 40 indivíduos saudáveis. Medimos a espessura do complexo de células maculares e ganglionares. Realizamos medições da espessura da coroide na região subfoveal e a 500 µm, 1000 µm e 1500 µm do centro da fóvea.
Resultados: Pacientes com retinite pigmentosa apresentaram espessuras maculares e da coroide significativamente mais finas em todas as medições e suas medidas individuais da espessura do complexo de células ganglionares foram inferiores às de indivíduos saudáveis. A espessura média do complexo de células ganglionares foi significativamente menor nos pacientes com retinite pigmentosa do que nos controles. A espessura macular média foi significativamente correlacionada com as espessuras médias do complexo das células de coroide e das células ganglionares médias. Não encontramoscorrelação entre a espessura media da coroide e a espessura media do complexo de células ganglionares.
Conclusões: A coroide foi levemente afetada em nossos pacientes com retinite pigmentosa precoce. A tendência à significância na retina interna foi possivelmente causada por uma boa acuidade visual.
Keywords: Coroide/anatomia & histologia; Retina/anatomia & histologia; Células ganglionares da retina; Retinite pigmentosa; Tomografia de coerência óptica
Arq. Bras. Oftalmol. 2021;84 (4 )
:367-373
| DOI: 10.5935/0004-2749.20210053
Abstract
OBJETIVO: A doença de Stargardt é a forma mais comum de distrofia macular de início juvenil. É bilateral e simétrica em aparência, afeta a mácula e sua característica principal é a diminuição da visão central que geralmente inicia-se na primeira ou segunda década de vida. O objetivo do estudo é descrever o perfil clínico dos pacientes avaliados no Complexo Hospital de Clínicas da Universidade Federal do Paraná, bem como descrever os achados eletrorretinográficos destes pacientes com o eletrorretinograma de campo total.
MÉTODOS: Foi realizado um estudo observacional retrospectivo, baseado na análise de prontuários e eletrorretinograma de 27 pacientes com Doença de Stargardt e Fundus Flavimaculatus, atendidos em consulta oftalmológica no ambulatório de Eletrofisiologia Ocular e Neuro-Oftalmologia do Complexo Hospital de Clínicas da Universidade Federal do Paraná, entre 1997 e 2014. Os pacientes incluídos no estudo apresentavam quadro clínico, fundoscopia e/ou achados eletrorretinográficos compatíveis com a doença.
RESULTADOS: A acuidade visual no melhor olho variou de 0 a 1,6 logMAR (20/20 a 20/800), com média de 0,89 ± 0,42 logMAR. A idade de aparecimento dos sintomas variou desde o nascimento a 36 anos (19,2 ± 9,2), sendo a maioria nas 1ª e 2ª década de vida. Em relação ao tempo entre o início dos sintomas e o diagnóstico, a média foi de 7,3 anos. Na fundoscopia, todos os pacientes apresentaram alguma alteração. Na análise do eletrorretinograma, a maioria dos pacientes demonstrou resultados que diferem da amostra de pacientes controles, ou seja, amplitudes reduzidas e tempos de culminação aumentados nas fases fotópicas e escotópicas.
CONCLUSÕES: A acuidade visual e idade de início de aparecimento dos sintomas encontrados neste estudo são compatíveis com a evolução desta distrofia. Achados fundoscópicos típicos da doença de Stargardt e eletrorretinograma alterados foram mais frequentes em decorrência do atraso no diagnóstico. Novos estudos prospectivos são necessários para avaliar estes pacientes, fundamentando-se em novas tecnologias.
Keywords: Eletrorretinografia; Doenças retinianas; Epitélio pigmentado da retina; Degeneração macular; Lipofuscina
Arq. Bras. Oftalmol. 2025;88 (3 )
:1-8
| DOI: 10.5935/0004-2749.2024-0104
Abstract
PURPOSE: This study aimed to characterize retinitis pigmentosa associated with the eyes shut homolog gene, which causes hereditary retinal degeneration.
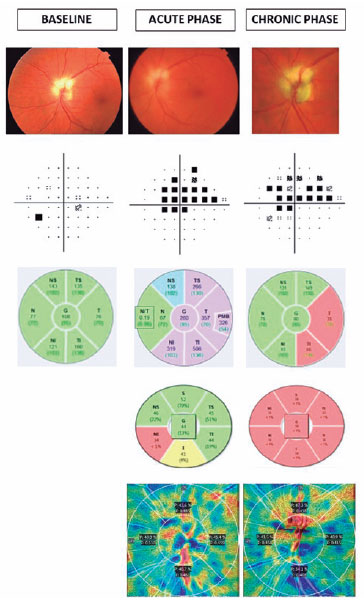
METHODS: The anatomical and functional findings of retinitis pigmentosa in patients with variants of the eyes shut homolog gene were characterized and compared using multimodal imaging and genetic analysis of the variants. Clinical data such as visual acuity, lens status, and refraction were obtained from medical records. Patients underwent an ophthalmic examination, including static visual field, microperimetry, optical coherence tomography, fundus autofluorescence, and fundus photography.
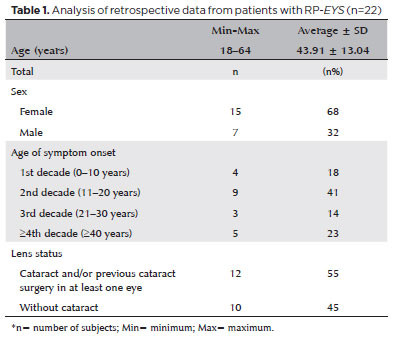
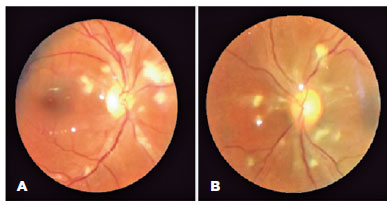
RESULTS: Twenty-two patients were included in the study. Several anatomical and functional characteristics of retinitis pigmentosa-eyes shut homolog were identified, including the presence of cataracts, cystoid macular edema, epiretinal membrane, and a tubular visual field. Genetic results revealed 26 distinct variants in the cohort, with 7 novel variants not previously documented or reported in the scientific literature or databases.
CONCLUSION: The findings demonstrate that eyes shut homolog-retinitis pigmentosa manifests in specific patterns, starting in adolescence with mild progression and advancing with age. The integration of multimodal imaging and genetic analysis has provided a detailed understanding of the anatomical and functional features of retinitis pigmentosa-eyes shut homolog. Seven novel variants of the eyes shut homolog gene have been identified. These findings enhance the understanding of eyes shut homolog-related retinitis pigmentosa characteristics of by detailing the spectrum of mutations in this gene within the Brazilian population.
Keywords: Retinal diseases/diagnostic imaging; Retinitis pigmentosa/genetics; Retinal degeneration; Eye proteins/genetics; Eye diseases, hereditary/genetics; Genes, recessive; Phenotype; Multimodal imaging; Tomography, optical coherence/methods; Fluorescein angiogr
Arq. Bras. Oftalmol. 2024;87 (2 )
:1-8
| DOI: 10.5935/0004-2749.2022-0319
Abstract
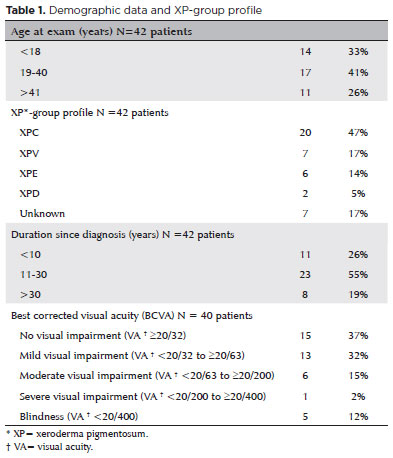
To assess Meibomian gland dysfunction using meibography in patients with xeroderma pigmentosum and correlate with ocular surface changes. This cross-sectional study evaluated patients with xeroderma pigmentosum. All patients underwent a comprehensive and standardized interview. The best-corrected visual acuity of each eye was determined. Detailed ophthalmic examination was conducted, including biomicroscopy examination of the ocular surface, Schirmer test type I, and meibography, and fundus examination was also performed when possible. Meibomian gland dysfunction was assessed by non-contact meibography using Oculus Keratograph® 5M (OCULUS Inc., Arlington, WA, USA). Saliva samples were collected using the Oragene DNA Self-collection kit (DNA Genotek Inc., Ottawa, Canada), and DNA was extracted as recommended by the manufacturer. Factors associated with abnormal meiboscores were assessed using generalized estimating equation models. A total of 42 participants were enrolled, and 27 patients underwent meibography. The meiboscore was abnormal in the upper eyelid in 8 (29.6%) patients and in the lower eyelid in 17 (62.9%). The likelihood of having abnormal meiboscores in the lower eyelid was 16.3 times greater than that in the upper eyelid.In the final multivariate model, age (p=0.001), mutation profile (p=0.006), and presence of ocular surface malignant tumor (OSMT) (p=0.014) remained significant for abnormal meiboscores. For a 1-year increase in age, the likelihood of abnormal meiboscores increased by 12%. Eyes with OSMT were 58.8 times more likely to have abnormal meiboscores than eyes without ocular surface malignant tumor.In the final model, age, xeroderma pigmentosum profile, previous cancer, and clinical alterations on the eyelid correlated with a meiboscore of ≥2.Meibomian gland dysfunction was common in patients with xeroderma pigmentosum, mainly in the lower eyelid. The severity of Meibomian gland dysfunction increases with age and is associated with severe eyelid changes.
Keywords: Meibomian glands/pathology; Meibomian glands/ diagnostic imaging; Photography; Xeroderma pigmentosum; Eyelid diseases/diagnostic imaging; Dry eye syndromes; DNA repair; Humans; Case report
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
09-fig01tb.jpg)
10-tab01tb.jpg)
02-fig01.jpg)
01-fig01.jpg)
03-fig01.jpg)
05-fig01.jpg)
02-fig01.jpg)
13-fig01.jpg)
14-fig01.jpg)
03-fig01.jpg)
15-fig01.jpg)