Arq. Bras. Oftalmol. 2006; 69 (3): 10.1590/S0004-27492006000300026
Total: 2639
Anna Paula Lemos de Albuquerque1; Jonathan Seiji Aguni2; Renato José Bett Correia2; Franciele Vegini2; André Dalsasso Borges de Souza2
DOI: 10.1590/S0004-27492006000300026
RESUMO
Os tumores linfóides dos anexos oculares são neoplasias de crescimento lento que acometem principalmente idosos. O objetivo deste trabalho é relatar um caso de linfoma não-Hodgkin de células da zona do manto, um subtipo raro de linfoma de células B nos anexos oculares. RELATO DO CASO: Paciente masculino de 62 anos com tumoração em pálpebras superiores, região maxilar e cavidade oral, tendo o diagnóstico inicial de doença de Mikulics, evoluiu com acentuado aumento das lesões, apesar da terapêutica imunossupressora. Biopsias da pálpebra superior esquerda e medula óssea revelaram linfoma não-Hodgkin de células B da zona do manto. Apesar da disseminação (estadiamento grau IV), quimioterapia e transplante de medula óssea conduziram à remissão da doença. COMENTÁRIOS CONCLUSIVOS: Apesar da disponibilidade de avançados métodos diagnósticos complementares como a imunofenotipagem e a análise genética molecular, o linfoma representa, para clínicos e patologistas, um desafio quanto ao diagnóstico e ao prognóstico.
Descritores: Linfoma de células do manto; Linfoma não-Hodgkin; Linfoma de células B; Antineoplásicos; Idoso; Relatos de casos [Tipo de publicação]
ABSTRACT
Ocular adnexal lymphoid tumors are slow-growing neoplasms that affect predominantly the elderly. The purpose of this study is to report a non-Hodgkin lymphoma from mantle zone cells, a rare ocular annexal B-cell lymphoma subtype. CASE REPORT: A 62-year-old male patient with superior eyelid, maxillary and oral tumours, had an initial diagnosis of Mikulics disease, developed lesion enlargement, despite immunosuppressant therapy. Left superior eyelid and bone marrow biopsy revealed non-Hodgkin lymphoma the mantle zone. In spite of dissemination (stage IV), chemotherapy and bone marrow transplant led to disease remission. CONCLUSIVE COMMENTARIES: Despite the availability of advanced complementary diagnostic methods, like immunophenotyping and molecular genetic analysis, lymphoma represents, for physicians and pathologists, a challenge regarding diagnosis and prognosis.
Keywords: Lymphoma, mantle-cell; Lymphoma, Non-Hodgkin; Lymphoma, B-cell; Aged; Case reports [Publication type]
INTRODUÇÃO
As lesões linfóides orbitais representam 10% de todos os tumores orbitais primários, enquanto os anexos orbitais podem ser envolvidos em cerca de 1,5% dos pacientes com linfoma não-Hodgkin sistêmico(1-2). É estimado ainda, que os linfomas dos anexos oculares representam aproximadamente 8% dos linfomas não-Hodgkin(3).
Os tumores linfóides dos anexos oculares são neoplasias de crescimento lento que, em geral, acometem idosos e se desenvolvem como manifestações primárias e secundárias na órbita, na conjuntiva e na pálpebra.
Tipicamente, pacientes com lesões linfóides orbitais têm uma massa visível ou palpável, proptose ou impedimento visual(3). A maioria destas lesões são tumores monoclonais de células B, mais precisamente, linfomas não-Hodgkin de células B da zona marginal, podendo incluir também linfomas difusos de grandes células B e linfomas foliculares. Subtipos menos comuns de linfomas de células B incluem: linfoma linfoplasmocítico, linfoma de células da zona do manto, plasmocitona e linfoma imunoblástico, em freqüência decrescente(4-10).
Nosso objetivo é apresentar um caso de linfoma de células B da zona do manto, raro subtipo de lesão linfóide dos anexos oculares.
RELATO DO CASO
JVG, masculino, 62 anos, auxiliar de enfermagem aposentado, natural e procedente de São José-SC, procurou o serviço de urgência oftalmológica do Hospital Regional de São José em abril de 2002. Queixava-se de ardência ocular, sensação de olhos e boca secos, além de dores musculares. Apresentava tumoração nas pálpebras superiores com quatro meses de evolução. Negava dores ou aumento de volume de articulações, perda de apetite ou de peso, lesões cutâneas ou sintomas respiratórios. Não tinha antecedentes familiares de tuberculose, doenças do colágeno ou doenças linfoproliferativas.
Apresentava na ocasião o seguinte exame: acuidade visual (AV) com correção de 20/30 em ambos os olhos (AO); à biomicroscopia apresentava catarata nuclear/subcapsular posterior de +/4 em AO; pressão intra-ocular e fundo de olho normais.
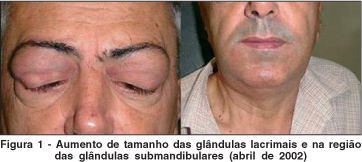
Ao exame externo foi encontrado aumento de volume difuso na topografia das glândulas lacrimais e das parótidas; nas regiões submandibular e sublingual e, inclusive, no palato (Figura 1). Tais massas apresentavam consistência fibro-elástica, eram pouco dolorosas à palpação e não demonstravam sinais flogísticos. As tumorações palpebrais estavam com uma dimensão aproximada de 3,5x3,5 cm. A motilidade ocular extrínseca se encontrava prejudicada pela presença de tais massas. O teste de Schirmer com e sem estímulo foi de 13 e 10 mm, respectivamente, e o tempo de ruptura do filme lacrimal foi de aproximadamente 8 segundos em AO. À coloração com rosa bengala, o paciente apresentou ceratite puntacta de ++/4 na metade inferior da córnea em AO e a conjuntiva se encontrava preservada.
A tomografia computadorizada (TC) axial demonstrou um aumento de volume, simétrico, de ambas as glândulas lacrimais, com limites definidos e valor de atenuação misto, apresentando em sua região central atenuação reduzida e borda com atenuação mais elevada, impregnando-se levemente pelo contraste (Figura 2). O laudo radiológico sugeriu diagnóstico de dacrioadenite crônica de causa a esclarecer. A radiografia de tórax, bem como hemograma e coagulograma estavam normais.

Outros exames laboratoriais apresentaram os seguintes resultados: VHS 22 mm 1ª hora; proteína C-reativa 6; glicemia 97 mg/dL; TSH 154; fator reumatóide inferior a 15; fator anti-nuclear negativo; anticorpos anti-Ro e anti-La (marcadores síndrome de Sjögren) negativos; VDRL e FTA-ABS não reagentes; PPD negativo; função hepática e renal normais. Foi, então, realizada biópsia incisional de glândulas salivares acessórias e de palato duro as quais demonstraram infiltração linfocítica difusa. Com base nestes achados, chegou-se ao diagnóstico mais provável de doença de Mikulicz (lesão linfoepitelial benigna), cujo tratamento consistiu de azatioprina, metotrexate e prednisona.
Em abril de 2004 apresentava oclusão dos eixos visuais pelas massas palpebrais, com cerca de 6 cm de diâmetro (Figura 3), e acuidade visual de conta dedos junto ao rosto (CDJR) em OD e de conta dedos a 2 m (CD 2 m) em OE. Foi constatada perda ponderal de 20 kg, relacionada à disfagia decorrente das massas na cavidade oral (Figura 3).

Avaliações histopatológica e imuno-histoquímica de biópsias palpebral (Figura 4) e da medula óssea revelou tratar-se de linfoma não-Hodgkin de células B, segundo a Classificação da Organização Mundial de Saúde. O estadiamento clínico foi determinado como de grau IV, com comprometimento predominantemente extra-nodal, da medula óssea, dos anexos oculares e da mucosa oral. Nesta época, o hemograma apresentava anemia e linfocitose.

Imunofenotipicamente a amostra da medula óssea revelou: CD5+, CD20+ CD23-, CD10-, antígeno leucocitário comum + e ciclina D1+. Este fenótipo indicou Linfoma não-Hodgkin de células B da zona do manto.
Quimioterapia com ciclofosfamida, vincristina e prednisona foi iniciada em junho de 2004, proporcionando, em uma semana, redução de cerca de 30% das massas na face e na região submandibular. Após a confirmação diagnóstica de linfoma não-Hodgkin de células B da zona do manto, realizou 4 ciclos de Hyper-CVAD [ciclofosfamida, vincristina, doxorrubicina (Adriamycin®) e dexametasona], evoluindo com ganho de peso, sem visceromegalias e com redução completa das massas na face e cavidade oral.
Em novembro de 2004 realizou transplante de medula óssea (transplante autogênico de células tronco hematopoéticas), apresentando-se com melhora significativa do quadro.
Na avaliação oftalmológica, em fevereiro de 2005 (Figura 5), as massas palpebrais, maxilares e orais haviam regredido completamente. A acuidade visual de CDJR em OD e de CD2m em OE era justificada pela presença de catarata total (OD) e nuclear e subcapsular posterior 3+/4+ (OE). A motilidade ocular extrínseca em AO e o mapeamento de retina em OE não revelaram alterações.

DISCUSSÃO
Os linfomas malignos representam um dos extremos do espectro das lesões linfoproliferativas, sendo o outro pólo representado pela hiperplasia linfóide reativa e o grupo intermediário denominado de hiperplasia linfóide atípica. Estas lesões são compostas de proliferações monoclonais ou policlonais de linfócitos. Tumores policlonais são geralmente considerados benignos, enquanto os monoclonais são tipicamente malignos(11). A maioria dos linfomas dos anexos oculares são tumores monoclonais de células B, quase exclusivamente, linfomas não-Hodgkin de células B(4,6-11).
Independente da entidade histopatológica (hiperplasia linfóide reativa, hiperplasia linfóide atípica ou linfoma maligno), pacientes com lesões linfoproliferativas dos anexos oculares não diferem significativamente quanto à idade, sexo, apresentação de sinais e sintomas ou achados oftálmicos(4,6,8). Com exceção da dor, que ocorre em tumores associados a erosões ósseas(4,6,12), há poucos sinais e sintomas que auxiliem na distinção clínica entre hiperplasia linfóide reativa e linfoma maligno.
As proliferações linfóides podem ocorrer em todas as idades, mas são mais comumente vistas em pacientes entre a 5ª e a 7ª décadas de vida(4-8,12). Tendem a afetar mais freqüentemente mulheres que homens (mulheres: homens = 1,5-2: 1). Ocorrem mais freqüentemente na órbita, seguido pela conjuntiva e as pálpebras(4-6,8,12). Aproximadamente 10 a 17% dos casos demonstram envolvimento bilateral com a bilateralidade ocorrendo simultaneamente em 80% dos casos. Sinais e sintomas típicos incluem massa orbital com ou sem proptose, diplopia, edema, hiperemia conjuntival (coloração salmão) e ptose quando há envolvimento da derme e do orbicular da pálpebra superior(4,6,8,12). Na época da avaliação inicial, nosso paciente tinha 60 anos e apresentava aumento de volume das pálpebras, das parótidas, das glândulas salivares e lacrimais. O acometimento destas últimas caracterizava massa de localização orbital. Entretanto, ao invés de proptose, observava-se ptose, a qual estava relacionada ao envolvimento palpebral. Dois anos mais tarde, houve até mesmo oclusão dos eixos visuais pelas massas palpebrais acompanhada de intensas hiperemia e edema conjuntivais.
Tomografia computadorizada, ressonância magnética e ecografia auxiliam na determinação do tamanho, número e grau da infiltração destas lesões. A maioria das lesões linfóides dos anexos oculares são massas homogêneas, unifocais, de densidade relativamente alta, geralmente se localizam dentro do tecido adiposo da órbita e tendem a se moldar às estruturas da órbita. Destruição óssea ou infiltração apenas são observadas em linfomas de alto grau(13). A tomografia realizada na avaliação inicial revelava aumento de volume homogêneo das glândulas lacrimais, de densidade elevada e sem sinais de lise óssea.
O diagnóstico definitivo é, geralmente, estabelecido através de exame histopatológico e imuno-histoquímico. Este último é utilizado para detectar imunoglobulinas na superfície dos linfócitos e determinar se uma população celular é policlonal ou monoclonal.
O exame histopatológico e a imunofenotipagem das biópsias realizadas apontaram para o diagnóstico de Linfoma não-Hodgkin de células da zona do manto (zona intermediária). Esta lesão é composta de linfócitos B pouco maiores que os linfócitos em estágio final de maturação e que apresentam irregularidades na superfície da membrana nuclear, como fendas, proeminentes indentações e pregas lineares. Corresponde a 5% de todos os linfomas não-Hodgkin(11,14).
Os cortes histológicos da biópsia removida do palato e das glândulas salivares acessórias revelaram infiltração linfocítica difusa, caracterizando uma lesão linfoepitelial benigna e conduzindo ao diagnóstico de doença de Mikulicz. No entanto, posterior reavaliação deste mesmo material demonstrou características histopatológicas de linfoma não-Hodgkin de células B. É possível que a abundância de tecido linfóide na orofaringe e a pequena representatividade do material da biopsia aliadas à heterogeneidade celular e à presença de centros germinativos reacionais, comuns em muitos desses tumores, tenham levado a uma falsa interpretação histopatológica.
É difícil predizer quais tumores permanecerão localizados e quais, eventualmente, progredirão para linfoma sistêmico. Burke e Jakobiec encontraram linfomas sistêmicos em pacientes com tumores imunofenotipicamente policlonais e, em contra-partida, observaram que o desenvolvimento de doença extra-ocular nos pacientes com tumores monoclonais era bastante raro(11,15).
A análise genética molecular estuda os padrões de rearranjos genéticos responsáveis pela produção de imunoglobulinas por células tumorais. Em geral, nas lesões policlonais há um número infinito de diferentes rearranjos genéticos, mas em um infiltrado monoclonal, todas as células do clone rearranjam seu DNA em um padrão idêntico. Entretanto, Johnson et al. estudando 77 pacientes com lesões linfóides oculares observaram que a análise genética molecular destes tumores não foi útil em predizer que pacientes desenvolveriam linfoma sistêmico(16). Na verdade, o estadiamento e a localização precisa da lesão possuem maior valor preditivo. Lesões conjuntivais têm a mais baixa incidência de linfoma extra-ocular; enquanto, lesão orbital apresenta comportamento intermediário e aquelas das pálpebras são as que mais freqüentemente evoluem com linfoma sistêmico(4,8,11,16). O paciente, em questão, possuía tumor monoclonal e localizado inicialmente nas órbitas e pálpebras. Apresentava, portanto, fatores prognósticos decisivos para o envolvimento sistêmico.
O tratamento dos linfomas dos anexos oculares sem qualquer evidência de doença extra-ocular é realizado através de radioterapia na dose de 24 a 50 Gy(4,17-18). Se doença sistêmica está presente, quimioterapia deve ser administrada. Com o esquema CHOP [ciclofosfamida, hidroxidoxorrubicina, vincristina (Oncovin®) e prednisona], indicado para a maioria desses tumores(4), houve resposta bastante satisfatória, caracterizada pela completa remissão das massas e significativa melhora clínica do paciente.
CONCLUSÃO
Apesar da disponibilidade de avançados métodos diagnósticos complementares como a imunofenotipagem e a análise genética molecular, o linfoma representa, para clínicos e patologistas, um desafio quanto ao diagnóstico e ao prognóstico. Na região ocular, outras entidades patológicas como o pseudotumor inflamatório e a hiperplasia linfóide reativa tornam esta questão ainda mais complexa. A avaliação histopatológica e o acompanhamento clínico cuidadosos são instrumentos fundamentais para a conduta terapêutica de todos os pacientes portadores de lesões linfóides dos anexos oculares, sejam elas benignas ou obviamente malignas.
Mesmo com disseminação, a maioria desses tumores apresenta comportamento indolente e o prognóstico de remissão a longo prazo é favorável.
REFERÊNCIAS
1. Jakobiec FA, Jones IS. Lymphomatous plasmacytic, histiocytic, and hematopoietic tumors. In: Jones IS, Jakobiec FA, editors. Disease of the orbit. Philadelphia: Harper & Row; 1981.
2. Wotherspoon AC, Diss TC, Pan LX, Schmid C, Kerr-Muir MG, Lea SH, Isaacson PG. Primary low-grade B-cell lymphoma of the conjunctiva: a mucosa-associated lymphoid tissue type lymphoma. Histopathology. 1993;23(5): 417-24.
3. Fung CY, Tarbell NJ, Lucarelli MJ, Goldberg SI, Linggood RM, Harris NL, Ferry JA. Ocular adnexal lymphoma: clinical behavior of distinct World Health Organization classification subtypes. Int J Radiat Oncol Biol Phys. 2003;57(5): 1382-91.
4. Coupland SE, Hummel M, Stein H. Ocular adnexal lymphomas: five case presentations and a review of the literature. Surv Ophthalmol. 2002;47(5):470-90. Review.
5. Auw-Haedrich C, Coupland SE, Kapp A, Schmitt-Graff A, Buchen R, Witschel H. Long term outcome of ocular adnexal lymphoma subtyped according to the REAL classification. Revised European and American Lymphoma. Br J Ophthalmol. 2001;85(1):63-9.
6. Coupland SE, Krause L, Delecluse HJ, Anagnostopoulos I, Foss HD, Hummel M, et al. Lymphoproliferative lesions of the ocular adnexa. Analysis of 112 cases. Ophthalmology. 1998;105(8):1430-41.
7. Jenkins C, Rose GE, Bunce C, Wright JE, Cree IA, Plowman N, et al. Histological features of ocular adnexal lymphoma (REAL classification) and their association with patient morbidity and survival. Br J Ophthalmol. 2000; 84(8):907-13.
8. Knowles DM, Jakobiec FA, McNally L, Burke JS. Lymphoid hyperplasia and malignant lymphoma occurring in the ocular adnexa (orbit, conjunctiva, and eyelids): a prospective multiparametric analysis of 108 cases during 1977 to 1987. Hum Pathol. 1990;21(9):959-73.
9. Medeiros LJ, Harris NL. Lymphoid infiltrates of the orbit and conjunctiva. A morphologic and immunophenotypic study of 99 cases. Am J Surg Pathol. 1989;13(6):459-71.
10. Nakata M, Matsuno Y, Katsumata N, Takenaka T, Kobayashi Y, Narabayashi M, et al. Histology according to the Revised European-American Lymphoma Classification significantly predicts the prognosis of ocular adnexal lymphoma. Leuk Lymphoma. 1999;32(5-6):533-43.
11. Jakobiec FA, Knowles DM. An overview of ocular adnexal lymphoid tumors. Trans Am Ophthalmol Soc. 1989;87:420-42; discussion 442-4.
12. White WL, Ferry JA, Harris NL, Grove AS Jr. Ocular adnexal lymphoma. A clinicopathologic study with identification of lymphomas of mucosa-associated lymphoid tissue type. Ophthalmology. 1995;102(12):1994-2006.
13. Flanders AE, Espinosa GA, Markiewicz DA, Howell DD. Orbital lymphoma. Role of CT and MRI. Radiol Clin North Am. 1987;25(3):601-13.
14. Weisenburger DD, Kim H, Rappaport H. Mantle-zone lymphoma: a follicular variant of intermediate lymphocytic lymphoma. Câncer. 1982;49(7):1429-38.
15. Burke JS. Histologic criteria for distinguishing between benign and malignant extranodal lymphoid infiltrates. Semin Diagn Pathol. 1985;2(3):152-62. Review.
16. Johnson TE, Tse DT, Byrne GE Jr, Restrepo A, Whitcomb CC, Voigt W, et al. Ocular adnexal lymphoid tumors: a clinicopathologic and molecular genetic study of 77 patients. Ophthal Plast Reconstr Surg. 1999;15(3):171-9.
17. Kennerdell JS, Flores NE, Hartsock RJ. Low-dose radiotherapy for lymphoid lesions of the orbit and ocular adnexa. Ophthal Plast Reconstr Surg. 1999; 15(2):129-33.
18. Mac Manus MP, Hoppe RT. Is radiotherapy curative for stage I and II low-grade follicular lymphoma? Results of a long-term follow-up study of patients treated at Stanford University. J Clin Oncol. 1996;14(4):1282-90.
Endereço para correspondência:
Anna Paula L. de Albuquerque
Rua Guilherme Weege, 50 - Sala 604
Jaraguá do Sul (SC) CEP 89251-610
Recebido para publicação em 19.09.2005
Aprovação em 04.01.2006
Trabalho realizado no Hospital Regional de São José - "Dr. Homero de Miranda Gomes" - São José (SC) - Brasil.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.