Arq. Bras. Oftalmol. 2005; 68 (6): 10.1590/S0004-27492005000600026
Total: 2907
Fabricio Witzel de Medeiros1; Rosana Aparecida Soares Altieri2; Murilo Barreto Souza3; José Antonio de Almeida Milani1; Milton Ruiz Alves1
DOI: 10.1590/S0004-27492005000600026
RESUMO
Ceratite intersticial não luética, surdez e vertigem caracterizam a síndrome de Cogan. Os achados oculares mais comuns no início da síndrome são opacidades corneanas numulares periféricas bilaterais. O tratamento desta rara doença utiliza imunossupressão sistêmica com agentes citotóxicos e corticosteróides. O relato de caso apresenta uma paciente com a evolução da forma clássica da síndrome de Cogan.
Descritores: Manifestações oculares; Síndrome; Vasculite; Surdez; Vertigem; Ceratite; Opacidade da córnea
ABSTRACT
Non-luetic interstitial keratitis, deafness and vertigo characterize Cogan's syndrome. The most common ocular findings in early Cogan's syndrome are bilateral, peripheral, subepithelial numular corneal opacities. The report presents a patient with the evolution of the classic form of Cogan's syndrome.
Keywords: Eye manifestations; Syndrome; Vasculitis; Deafness; Vertigo; keratitis; Corneal opacity
INTRODUÇÃO
A síndrome de Cogan, inicialmente descrita por D. G. Cogan em 1945(1) em sua forma clássica constitui uma doença rara de provável etiologia autoimune caracterizada por inflamação ocular recorrente, sintomas vestíbulo-auditivos e perda auditiva do tipo neurossensorial. A forma clássica da doença é caracterizada pela apresentação de uma ceratite intersticial não sifilítica associada à disfunção vestíbulo-auditiva como na doença de Menière. Em sua evolução, leva à perda da acuidade auditiva em poucos meses, perda esta que pode ser permanente caso o diagnóstico seja retardado privando o paciente do tratamento. A forma atípica geralmente envolve toda a estrutura ocular levando a episódios recorrentes de conjuntivites, episclerites, uveítes, edema de disco óptico e vasculite retiniana(2-5). Além disso, a doença inflamatória vascular generalizada é mais comum nessa forma da doença, gerando um pior prognóstico do ponto de vista sistêmico(3,6-7).
RELATO DE CASO
Trata-se de uma mulher de 28 anos com história de perda da acuidade auditiva e zumbido desde setembro de 2003, associado ao aparecimento de lesões esbranquiçadas corneanas em ambos os olhos de maneira progressiva. A paciente refere hiperemia conjuntival acompanhando o quadro, nega dor ocular e relata discreta fotofobia.
Refere o uso irregular de colírios a base de dexametasona a 0,1% prescritos em outros serviços como única forma de tratamento, além do otofone.
No exame oftalmológico mediu-se a acuidade visual com correção de 20/20 em ambos os olhos. Reflexos fotomotores presentes e simétricos bilateramente. Motilidade ocular extrínseca sem alterações.
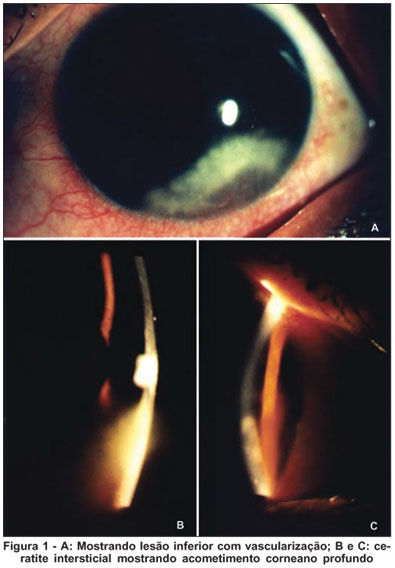
Na biomicroscopia observava-se infiltrados corneanos intersticiais bilateralmente em média-periferia poupando o eixo visual, associados a hiperemia conjuntival reacional e vascularização corneana profunda relacionada às lesões (Figura 1). Nota-se ainda reação de câmara anterior 1+/4+ em ambos os olhos. O exame de fundo de olho foi considerado normal, não evidenciando sinais de vasculite ou edema do nervo óptico em ambos os olhos.
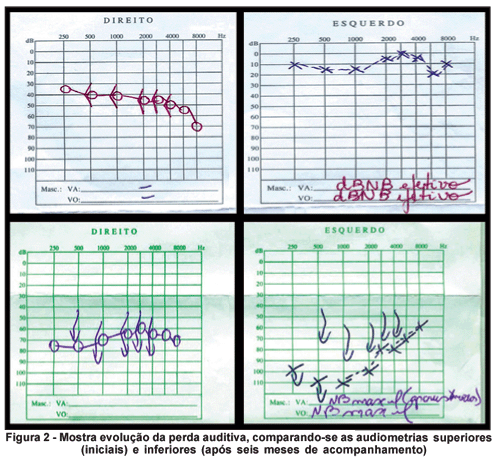
Nos exames laboratoriais a paciente apresentou sorologias negativas para clamídia e sífilis (VDRL e FTABS), hemograma normal, radiografia de tórax normal, audiometria evidenciou perda auditiva neurosensorial bilateralmente (Figura 2), potenciais evocados auditivos de tronco encefálico (BERA) dentro da normalidade, anticorpos anti-cardiolipina negativos (IgG e IgM) negativos, anticorpos anti-citoplasma (ANCA) negativos (P e C ANCA), anticorpos anti-ENA (RNP) não reagente, complemento C4 abaixo dos valores de referência, fator anti-núcleo negativo, complemento C3 acima do normal (214,3 mg/dl; referência de 90 a 180 mg/dl), complemento total normal, anti-HIV 1 e 2 negativos, teste tuberculínico (PPD) de 10 mm, VHS de 40 mm (normal até 20 mm), tomografia de ossos temporais normal, sorologia para Herpes I e II (IgG positivo e IgM negativo).
A paciente recebeu esquema imunossupressor inicial com prednisona 1,3mg/kg/dia, metotrexate 0,18 mg/kg/semana, além de reposição de carbonato de cálcio (1250 mg 1 x/semana), calciferol (vitamina D) na dose de 500 UI/dia e ácido fólico (5 mg/semana). Recebeu também dexametasona 0,1% tópica 4 x/dia e tropicamida 2 x/dia para tratamento do quadro inflamatório ocular.
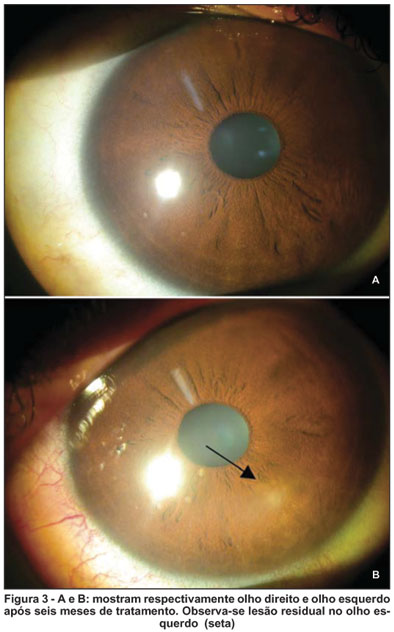
Houve boa evolução do quadro com desaparecimento das lesões no olho direito e diminuição importante destas no olho esquerdo (Figura 3) nestes últimos 6 meses. A paciente evoluiu com hipertensão ocular após introdução do colírio de corticóide e, portanto, este foi suspenso e introduzida medicação tópica antihipertensiva com resolução do quadro.
DISCUSSÃO
Acredita-se que a síndrome de Cogan contemple um fundo de autoimunidade em sua origem sendo associada a outras doenças que apresentam essa mesma característica (doença de Wegener, poliarterite nodosa, artrite reumatóide)(8). A forma atípica da doença está mais relacionada aos comemorativos sistêmicos, sendo mais agressiva e de pior prognóstico(3,6-7). Os sintomas oculares da forma atípica (conjuntivite, episclerite, edema de disco óptico, vasculites retinianas) podem preceder em anos os sintomas vestíbulo-auditivos, dificultando o diagnóstico da doença.
O quadro apresentado se encaixa dentro do diagnóstico da forma clássica da síndrome de Cogan, que faz parte do diagnóstico diferencial das ceratites intersticiais não luéticas. A tuberculose e a moléstia de Hansen devem estar também como possíveis suspeitas diagnósticas. A ceratite, associada à perda auditiva do tipo neurossensorial, é muito característica da doença(1), embora os sintomas vestíbulo-auditivos sejam praticamente indistinguíveis da doença de Menière.
Os corticosteróides tópicos se prestam ao tratamento dos sinais inflamatórios oculares embora os efeitos dessas drogas nesses pacientes permaneçam incertos(2). Isto deve ser levado em consideração na ilustração desse caso uma vez que a paciente desenvolveu glaucoma cortisônico secundário ao tratamento.
Vale ressaltar o tempo do início dos sintomas até o diagnóstico, sendo que em pacientes com processos inflamatórios oculares recorrentes ou de evolução prolongada, os sintomas vestibulares e auditivos devem ser investigados, uma vez que o prejuízo dessas funções está diretamente ligado ao início da terapia imunossupressora que deve ser o mais precoce possível.
A síndrome de Cogan torna-se um modelo claro de que a intervenção multidisciplinar otimiza o diagnóstico precoce influindo diretamente no prognóstico da doença(9).
REFERÊNCIAS
1. Cogan DG. Syndrome of non syphilitic interstitial keratitis and vestibulo-auditory symptoms. Arch Ophthalmol. 1945;33:144-9.
2. Haynes BF, Kaiser-Kupfer MI, Mason P, Fauci AS. Cogan syndrome: studies in thirteen patients, long-term follow-up, and a review of the literature. Medicine (Baltimore). 1980;59(6):426-41.
3. Bennet FM. Bilateral recurrent episcleritis associated with posterior corneal changes, vestibulo-auditory symptoms rheumatoid arthritis. Am J Ophthalmol. 1963;55:815-8.
4. Peeters GJ, Cremers CW, Pinkers AJ, Hoefnagels WH. Atypical Cogan's syndrome: and autoimmune disease? Ann Otol Rhinol Laryngol. 1986;95(2 Pt 1): 173-5.
5. Shah P, Luqmani RA, Murray PI, Honan WP, Corridan PG, Emery P. Posterior scleritis - an unusual manifestation of Cogan's syndrome. Br J Rheumatol. 1994;33(8):774-5.
6. LaRaja RD. Cogan syndrome associated with mesenteric vascular insufficiency. Arch Surg. 1976;111(9):1028-31.
7. Bicknell JM, Holland JV. Neurologic manifestations of Cogan syndrome. Neurology. 1978;28(3):278-81.
8. Lunardi C, Bason C, Leandri M, Navone R, Lestani M, Millo E, et al. Autoantibodies to inner ear and endothelial antigens in Cogan's syndrome. Lancet.2002;360(9337):915-21.
9. Orsoni JG, Zavota L, Pellistri I, Piazza F, Cimino L. Cogan syndrome. Cornea. 2002;21(4):356-9.
Endereço para correspondência
Rua Alves Guimarães, 856 - Apto. 32
São Paulo (SP) CEP 05410-001
Recebido para publicação em 04.04.2005
Versão revisada recebida em 02.08.2005
Aprovação em 15.08.2005
Trabalho desenvolvido junto ao Departamento de Oftalmologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (USP); em parceria com o Departamento de Reumatologia da Universidade Federal de São Paulo (UNIFESP).
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência do Dr. Ítalo Mundialino Marcon sobre a divulgação de seu nome como revisor, agradecemos sua participação neste processo.
Nenhum dos autores tem interesse financeiro relacionado ao trabalho.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.