Arq. Bras. Oftalmol. 2000; 63 (2): 10.1590/S0004-27492000000200012
Total: 2784
Rosália M. S. Antunes-Foschini1; Harley E. A. Bicas2
DOI: 10.1590/S0004-27492000000200012
RESUMO
Objetivo: Descrição de quadro clínico de criança com síndrome de Opitz, estrabismo e outros distúrbios oculomotores. Relato de caso: Paciente de 3 anos com hipospádia peno-escrotal, escroto bífido, ânus anteriorizado, micrognatia, pálato alto, fronte ampla, raiz nasal baixa, hipertelorismo e epicanto, implantação baixa de orelhas, clinodactilia, espinha bífida e retardo do desenvolvimento neuromotor, interpretadas como manifestações da síndrome de Opitz (OMIM *145410). Apresentava uma ET alternante com fixação cruzada (síndrome de Ciancia), associada a manifestações da síndrome de Duane bilateral e desvio vertical (E/D), possivelmente devido a uma dissociação motora entre o RSD e músculos palpebrais do OD. A criança faleceu após cirurgia para reconstrução anorretal. Discussão: Apesar de comprometimentos oculares, não há publicações referentes à síndrome de Opitz em periódicos oftalmológicos. As com menções sobre estrabismo não são acompanhadas por relatos mais pormenorizados da motilidade ocular. O caso apresentava dissociações oculomotoras importantes, mas seus esclarecimentos não foram possíveis pelo falecimento prematuro da criança.
Descritores: Síndrome de Opitz; Estrabismo
ABSTRACT
Objetive: Description of a clinical case of a child presenting Opitz syndrome, strabismus and other oculomotor disturbances. Case report: A 3-year-old child, with penoscrotal hypos-padia, bifid scrotum, micrognathy, high palate, proeminent forehead, low nasal bridge, hypertelorism, epicanthus, low-set ears, clinodactyly, bifid spine, delayed motor develop-ment and anteriorly placed anus as manifestations of the Opitz syndrome (OMIM *145410). He presented also an alternating esotropia with crossed fixation (Ciancia's syndrome), associated with manifestations of a bilateral Duane's syndrome and a vertical deviation (L/R), possibly due to dissociated actions between the RSR and the palpebral muscles of the right eye. The child died after a surgery for an anorectal reconstruction. Discussion: Although presenting ocular manifestations, there are no papers referring to the Opitz syndrome in ophthalmological publications. Those which mention strabismus have no proper descriptions of the ocular motility conditions. The present case showed important oculomotor dissociations, but their causes could not be clarified, due to the premature death of the child.
Keywords: Opitz syndrome; Strabismus
RELATOS CURTOS
Síndrome de Opitz - relato de caso
Opitz syndrome - a case report
Rosália M. S. Antunes-Foschini 1
Harley E. A. Bicas 2
INTRODUÇÃO
A síndrome de Opitz (OMIM *145410)1 ou, ainda, síndrome de Hipertelorismo-Hipospádia, ou síndrome de Opitz-Frias, ou síndrome óculo-genito-laringeal de Opitz foi inicialmente descrita em 1969 por Opitz e colaboradores 2, 3. Primeiramente ela foi subdividida em duas outras distintas, a síndrome G, assim designada por ter sido descrita em uma família cujo nome tinha por inicial a letra G e que possuía quatro irmãos acometidos 3; e a síndrome BBB, descrita em três famílias diferentes, todas com nomes iniciados pela letra B, que possuíam oito homens acometidos 2. Posteriormente, chegou-se à conclusão que as síndromes G e BBB faziam parte da mesma condição 4-6, mas ainda há autores que a subdividem em duas entidades diferentes 7, 8. Ainda não se conhece sua forma de transmissão genética, mas a herança deve ser autossômica dominante ou ligada ao X. Recentemente, estudos genéticos moleculares têm demonstrado que ela representa uma desordem heterogênea localizada no cromossomo 22 na região q 11.2 9,10 e no cromossomo X na região p 22 11, 12.
Caracteriza-se por defeitos no plano mediano do corpo, sobretudo no esqueleto craniofacial e no tubérculo genital. Seus principais elementos de diagnóstico são a presença de telecanto e de hipospádia no sexo masculino; e, no feminino, de telecanto e acometimento de hipospádia em parentes do sexo masculino 2-8.
Sobre as ocorrências relativas das anomalias comuns à síndrome, há diferentes publicações, algumas referindo-as, outras não. Por exemplo, Buyse 8, ao descrever a síndrome BBB, adianta: telecanto (ou aparente hipertelorismo) e hipospádia em 97% dos casos; assimetria cranial (plagiocefalia) em 47%; retardo mental em 43%; estrabismo em 37%; criptorquidia em 30%; cardiopatia congênita em 25%; fenda labial e palatina em 23%; anomalias do trato urinário em 19% e ânus imperfurado em 5%. Mas também são ainda referidas as manifestações de rimas palpebrais oblíquas, pregas epicânticas, ponte nasal plana e larga com narinas antevertidas, frênulo da língua curto, rotação posterior das orelhas, micrognatia, escroto bífido, hérnias, sulcos na ponta do nariz, lábio superior fino, úvula bífida, língua fendida, anomalias dentárias, agenesia ou hipoplasia de corpo caloso, hipoplasia do vermis cerebelar, atrofia cortical, macro cisterna magna, grande "cavum septum pellucidum", fendas laringotraqueais, malformações na laringe, fístulas traqueoesofágicas, epiglote hipoplásica, carina alta, hipoplasia pulmonar, estenose duodenal, diástase de retos e hipotonia.6 O estrabismo é apenas citado em algumas publicações 6-8. Sua prevalência é indeterminada. Para a síndrome BBB, há acometimento de 37 homens para cada 31 mulheres, conforme os casos já relatados 7. Não foram encontrados dados sobre prevalência em diferentes raças.
RELATO DE CASO
MAC, 3 anos, masculino, procedente de Itápolis, SP, com história de esotropia (ET) e olho direito (OD) mais fechado, desde o nascimento. Nos antecedentes pessoais, diagnóstico de polidrâmnio no sétimo mês de gestação. Nos antecedentes familiares, nada digno de nota.
Ao exame ocular externo, rima palpebral direita menor, com distância do centro da pupila à margem palpebral superior igual a 2 mm à direita e 4 mm à esquerda. Posição viciosa de cabeça (figura 1a) e fixação cruzada (figura 1b e c). Uso de musculatura frontal para aumentar a rima palpebral D. Esotropia com fixação cruzada. Hipertelorismo. Epicanto (figura 1).
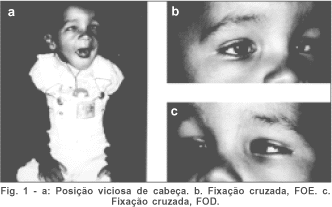
Ao teste de cobertura, confirma-se esotropia alternante e desvio vertical E/D; boa fixação com cada olho, mas preferência pelo esquerdo. Na fixação com esse olho (OE), em frente, pseudoblefaroptose D (figura 2c); uma pseudoblefaroptose E também ocorre na fixação com o OD, mas é menor (figura 2d). Desvio horizontal (ET) avaliado pelo método de Krimsky (pelas dificuldades de medida com o teste de cobertura) em torno de 50D. O desvio vertical E/D foi estimado em torno de 15°.
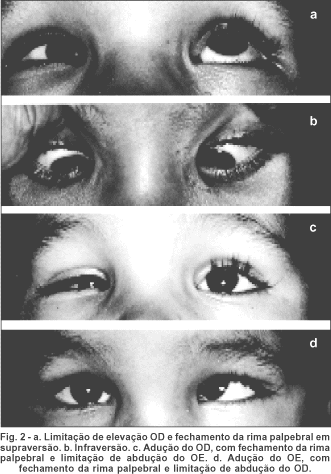
Ao exame das rotações oculares, limitação bilateral de abdução absoluta (olhos não passavam da linha média). OD também com limitação de elevação (figura 2a). Assim, o desvio horizontal mantinha-se aproximadamente simétrico em lateroversões, mas o vertical aumentava no olhar para cima. Fechamento da rima palpebral na adução do OD (figura 2c) e do OE (figura 2d) e na supra (figura 2a) e infradução do OD.
Na refratometria sob cicloplegia, OD = +0,5 D esf e OE = +1,5 D esf. À biomicroscopia, exame sem anormalidades. À oftalmoscopia: pólos posteriores sem alterações.
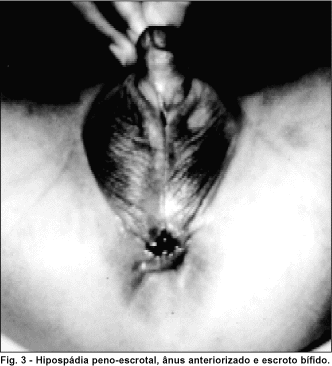
No exame clínico geral foram detectadas: hipospádia peno-escrotal, ânus anteriorizado e escroto bífido (figura 3), micrognatia, pálato alto, fronte ampla, raiz nasal baixa, implantação baixa de orelhas (figura 1a), clinodactilia bilateral discreta de quinto dedo das mãos, espinha bífida em coluna torácica e retardo de desenvolvimento neuromotor.
Foi indicada e realizada cirurgia de anorretoplastia. A operação teve bom resultado, mas no terceiro dia pós-operatório a criança apresentou náuseas, seguidas de crise de tosse seca, "desvio ocular", cianose e parada respiratória. Foram feitas manobras de ressuscitação cárdio-respiratória, inefetivas. A causa da morte não ficou definida nem mesmo pela necrópsia.
DISCUSSÃO
Não foram encontrados relatos da síndrome de Opitz no Brasil e, apesar do acometimento ocular, também não aparecem publicações dela em periódicos de Oftalmologia. As publicações sobre síndrome de Opitz, mencionando estrabismo, não descrevem outras informações sobre a motilidade ocular dos portadores.
O fenótipo descrito (síndrome de Opitz OMIM *145410)1 tem herança autossômica dominante, mapeia para o cromossomo 22q11.2, mas ainda não tem gene identificado 13-15.
Na presente descrição, as alterações da motilidade ocular externa não foram acompanhadas por outras anomalias oculares. Os achados de ET alternante com fixação cruzada e limitação bilateral de abdução são comuns em crianças com Síndrome de Ciancia 16; já o fechamento da rima palpebral às tentativas de adução (no caso, tanto do OD como do OE) são características da síndrome de Duane 17, do tipo I de Huber 18. Mas não se pode descartar a possibilidade de uma paralisia completa e bilateral dos nervos motores oculares externos (VI par cranial), com perda da elasticidade dos músculos retoslaterais e, pois, restrição de movimentos oculares no sentido oposto aos da ação deles, explicando a enoftalmia às tentativas de adução e a conseqüente diminuição da rima palpebral nessas circunstâncias. Aliás, essa etiopatogenia da síndrome de Duane (fibrose dos retos laterais)19 era a que prevalecia até a comprovação da disfunção inervacional dos retos laterais, demonstrada por eletromiografias 20.
O desvio vertical E/D poderia ser devido a uma paresia do músculo reto superior D (hipotropia do OD na fixação com o OE); ou a uma paresia do músculo reto inferior E (hipertropia do OE na fixação com o OD). O fechamento palpebral, ou pseudoblefaroptose assimétrica, maior à direita, pode, aliás, ser explicado pelo desvio vertical (hipotropia D) associado. A primeira hipótese talvez fosse mais provável, pois é compatível com uma possível inervação anômala dos músculos retos verticais do OD e dissociações de suas ações com as dos músculos para movimentos das pálpebras, visto observar-se diminuição da rima palpebral desse olho às tentativas de sua elevação ou abaixamento. Infelizmente, após o primeiro exame da movimentação ocular no qual a baixa colaboração da criança não permitiu maiores avanços na elucidação diagnóstica, deu-se o falecimento dela, ficando inconclusas as hipóteses levantadas.
SUMMARY
Objetive: Description of a clinical case of a child presenting Opitz syndrome, strabismus and other oculomotor disturbances. Case report: A 3-year-old child, with penoscrotal hypos-padia, bifid scrotum, micrognathy, high palate, proeminent forehead, low nasal bridge, hypertelorism, epicanthus, low-set ears, clinodactyly, bifid spine, delayed motor develop-ment and anteriorly placed anus as manifestations of the Opitz syndrome (OMIM *145410). He presented also an alternating esotropia with crossed fixation (Ciancia's syndrome), associated with manifestations of a bilateral Duane's syndrome and a vertical deviation (L/R), possibly due to dissociated actions between the RSR and the palpebral muscles of the right eye. The child died after a surgery for an anorectal reconstruction. Discussion: Although presenting ocular manifestations, there are no papers referring to the Opitz syndrome in ophthalmological publications. Those which mention strabismus have no proper descriptions of the ocular motility conditions. The present case showed important oculomotor dissociations, but their causes could not be clarified, due to the premature death of the child.
Keywords: Opitz syndrome; Strabismus.
REFERÊNCIAS BIBLIOGRÁFICAS
1. NCBI - National Center for Biotechnology Information. OMIM-Online Mendelian Inheritance in Man. Disponível em <http://www.ncbi.nlm.nih.gov/omim>
2. Opitz JM, Summmitt RL, Smith DW. The BBB syndrome. Familial telecanthus with associated congenital anomalies. In: Bergsma D ed., The Clinical Delineation of Birth Defects. Part II. Malformation Syndromes. New York: The National Foundation, 1969; BD:OAS 5:86-94.
3. Opitz JM, Frias JL, Gutenberger JE, Pellett JR. The G syndrome of multiple congenital anomalies. In: Bergsma D ed. The Clinical Delineation of Birth Defects. Part II. Malformation Syndromes. New York: The National Foundation, 1969; BD:OAS 5:95-101.
4. Cordero JF, Holmes, LB. Phenotypic overlap of the BBB and G syndromes. Am J Med Genet 1978;2(2):145-52.
5. Cappa M, Borrelli P, Marini R, Neri G. The Opitz Syndrome: a new designation for the clinically indistinguishable BBB and G Syndromes. Am J Med Genet 1987;28(2):303-9.
6. Jones KL. Opitz Syndrome. In: Jones KL ed., Smith's Recognizable Patterns of Human Malformation. Philadelphia: W.B.Saunders Company, 1997; cap.1, 132-7.
7. Buyse ML. Hypertelorism-Hypospadias Syndrome. In: Buyse ML ed., Birth defects Encyclopedia. Dover: Center for Birth Defects Information Services, Inc., 1990;p. 912-4.
8. Buyse ML. G Syndrome. In: Buyse ML ed., Birth defects Encyclopedia. Dover: Center for Birth Defects Information Services, inc., 1990; p. 755-7.
9. Fryburg JS, Lin KY, Golden WL. Chromosome 22q11.2 Deletion in a Boy With Opitz(G/BBB) Syndrome. Am J Med Genet 1996;62:274-5.
10. Robin NH, Feldman GJ, Aronson AL, Mitchell HF, Weksberg R, Leonard CO, Burton BK, Josephson KD, Laxova R, Aleck KA, Allanson JE, Guion-Almeida ML, Martin RA, Leichtman LG, Price RA, Opitz JM, Muenke M. Opitz syndrome is genetically heterogeneous, with one locus on Xp22, and a second locus on 22q11.2. Nat Genet 1995;11:459-61.
11. Quaderi NA, Schweiger S, Gaudenz K, Franco B, Rugarli EI, Berger W, Feldman GJ, Volta M, Andolfi G, Gilgenkrantz S, Marion RW, Hennekam RC, Opitz JM, Muenke M, Ropers HH, Ballabio A. Opitz G/BBB syndrome, a defect of Midline development, is due to mutations in a new RING finger gene on Xp22. Nat Genet 1997;17(3):285-91.
12. Dal Zotto L, Quaderi NA, Elliott R, Lingerfelter PA, Carrel L, Valsecchi V, Montini E, Yen C-H, Chapman V, Kalcheva I, Arrigo G, Zuffardi O, Thomas S, Willard HF, Ballabio A, Disteche CM, Rugarli EI. The mouse Mid1 gene: implications for the pathogenesis of Opitz syndrome and the evolution of the mammalian pseudoautosomal region. Hum Molec Genet 1998;7:489-99.
13. Lacassie Y, Arriaza MI. Opitz GBBB syndrome and the 22q11.2 deletion. (Letter) Am J Med Genet 1996;62:318.
14. McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J, Paris Y, Weinberg P, Clark BJ III, Emanuel BS, Zackai EH. Autosomal dominant `Opitz"GBBB syndrome due to a 22q11.2 deletion. AM J Med Genet 1995;59:103-13.
15. Robin NH, Opitz JM, Muenke M. Opitz G/BBB syndrome: clinical comparisons of families linked to Xp22 and 22q, and a review of the literature. Am J Med Genet 1996;62:305-17.
16. Ciancia AO. La esotropia com limitación de la abducción en el lactante. Arch Oftalmol B Aires 1962;26:207.
17. Duane A. Congenital deficiency of abduction, associated with impairment of adduction, retraction movements, contraction of the palpebral fissure and oblique movements of the eye. Arch Ophtalmol 1905;34:133.
18. Huber A. Duane's retraction syndrome: considerations on pathogenesis and aethiology of the different forms of Duane's retraction syndrome. In Strabismus 69. Henry Kimpton, London, 1970; p.36-43.
19. Türk S. Ueber Retractionsbewegungen der Augen Dtsch Med Wschr 1896; 22:199-201.
20. Breinin GM. Electromiography: a tool in ocular and neurologic diagnosis: II Muscles palsies. Arch Ophtalmol 1957;57:165-75.
Trabalho realizado no Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da USP.
1 Médica assistente junto ao Setor de Estrabismo e Ortóptica do Hospital das Clínicas da FMRP.
2 Professor Titular, Departamento de Oftalmologia e Otorrinolaringologia da FMRP.
Endereço para correspondência: Departamento de Oftalmologia, HCFMRP. Ribeirão Preto (SP) Brasil. CEP 14048-900.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.