Arq. Bras. Oftalmol. 2004; 67 (4): 10.1590/S0004-27492004000400022
Total: 1929
Namir Clementino Santos1; Luciene Barbosa de Sousa3; Virginia Fernandes Moça Trevisani1; Denise de Freitas1; Luis Antonio Vieira3
DOI: 10.1590/S0004-27492004000400022
RESUMO
OBJETIVO: Estudar as características clínicas, tratamento e evolução de pacientes com acometimento da córnea e esclera associados a doenças do tecido conectivo. MÉTODO: Descrição das alterações de segmento anterior em nove pacientes com doenças do tecido conectivo, previamente diagnosticada (5 casos) ou no qual o acometimento ocular foi sua primeira manifestação (4 casos). Todos os pacientes foram atendidos no setor de Doenças Externas Oculares e Córnea da Universidade Federal de São Paulo - Escola Paulista de Medicina (UNIFESP-EPM), acompanhados no ambulatório no período de julho/1999-dezembro/2000 e submetidos a exame oftalmológico completo, avaliação sistêmica e investigação laboratorial. Tratamento clínico e/ou cirúrgico foi proposto de acordo com a gravidade e evolução das manifestações oculares. RESULTADOS: Os diagnósticos sistêmicos observados nos portadores de doença inflamatória do segmento anterior foram de artrite reumatóide em sete pacientes (77,8%), esclerose sistêmica e granulomatose de Wegener em um paciente cada (22,2%). As manifestações oculares mais freqüentes foram esclerite (66,6%), ceratite ulcerativa periférica (55,5%) e ceratoconjuntivite seca (44,4%). Oitenta e nove por cento dos pacientes necessitaram de terapia imunossupressora sistêmica para o controle da inflamação ocular. A remissão da inflamação observou-se no período de três meses após o início do imunos-supressor (metotrexato, ciclofosfamida e/ou ciclosporina A). Em 55,5% dos pacientes uma abordagem cirúrgica (ressecção da conjuntiva do limbo, adesivo tecidual com lente de contato terapêutica, enxerto de esclera, transplante de córnea) se fez necessária. CONCLUSÃO: O envolvimento da córnea e da esclera associado à doença do tecido conectivo é sinal de atividade da doença e geralmente necessita de terapia imunossupressora para o seu controle. O oftalmologista deve estar alerta para detecção precoce e tratamento apropriado da doença de base.
Descritores: Esclerite; Esclerite; Úlcera de córnea; Úlcera de córnea; Tecido conjuntivo; Imunossupressão; Relato de caso
ABSTRACT
PURPOSE: To study the clinical features, diagnosis, treatment, and outcome of nine patients with corneal and scleral involvement associated to connective tissue disease. METHODS: Description of anterior segment abnormalities observed in nine patients with connective tissue disease, five of them previously diagnosed and four with the ocular involvement being the first manifestation of the disease. All patients were evaluated at the Corneal and External Diseases outpatient clinic of the Federal University of São Paulo - Escola Paulista de Medicina (UNIFESP-EPM) between July/1999 to December/2000 and were submitted to a complete ophthalmologic examination, clinical evaluation and laboratory investigation. Clinical or surgical treatment was indicated according to the ocular disease evolution. RESULTS: The clinical diagnoses of the patients with anterior segment inflammatory disease were rheumatoid arthritis in seven patients (77.8%), systemic sclerosis and Wegener´s granulomatosis in each of the remaining patients (22.2%). The most frequent ocular manifestation was scleritis (66.6%), followed by peripheral ulcerative keratitis (55.5%) and dry eye syndrome (44.4%). Eighty-nine percent (89%) of the patients required immunosuppressive therapy to control the ocular inflammatory disease. Remission of the ocular symptoms were observed after 3 months of the beginning of imunossupressive therapy on average. In 55.5% of the patients a surgical approach (conjunctival resection, application of cyanoacrylate tissue adhesive, penetrating keratoplasty and scleral patch graft) was necessary. CONCLUSION: The finding of corneal and scleral involvement associated with connective tissue disease is a sign of the disease activity and usually requires imunossupressive therapy. The ophthalmologists should be aware of these conditions in order to early diagnose and apply the appropriate treatment.
Keywords: Scleritis; Scleritis; Corneal ulcer; Corneal ulcer; Connective tissue; Immunossupression; Case report
INTRODUÇÃO
As doenças inflamatórias da periferia da córnea e da esclera, representada principalmente pela ceratite ulcerativa periférica, podem se apresentar de várias formas, de episódios autolimitados a processos rapidamente progressivos e destrutivos. Elas assumem papel importante pela freqüente associação com doenças sistêmicas potencialmente letais(1-6).
Diante do quadro de ceratite ulcerativa periférica é importante estabelecer o diagnóstico diferencial com as causas infecciosas e as doenças auto-imunes (doenças do tecido conectivo e úlceras de Mooren)(1). Qualquer infecção ocular pode estar associada com ulceração da periferia da córnea. Infecções sistêmicas também estão associadas com ulceração da periferia da córnea, tais como infecções por Neisseria, a tuberculose, a sífilis e as infecções pelo vírus da imunodeficiência humana(1). A úlcera de Mooren, por sua vez, é reconhecida como uma rara doença inflamatória de provável etiologia auto-imune caracterizada por ulceração da periferia da córnea. Seus achados clínicos podem ser indistinguíveis daqueles da ceratite ulcerativa periférica associada às doenças do tecido conectivo. Entretanto, a úlcera de Mooren é, por definição, idiopática e, portanto, não pode ser associada com qualquer doença sistêmica que possa contribuir para doença corneana(1).
A maioria das doenças do tecido conectivo pode produzir manifestações oculares, entretanto, quando ceratite ulcerativa ocorre, especial atenção deve ser dada ao diagnóstico de um grupo de doenças que inclui artrite reumatóide, granulomatose de Wegener, policondrite recorrente, poliarterite nodosa e lupus eritematoso sistêmico(1-2,4).
Outras manifestações oculares comuns a este grupo de doenças são ceratoconjuntivite seca, episclerite, esclerite e vasculite da coróide e da retina(1-6). Todas estas manifestações podem ocorrer paralelamente à atividade da doença sistêmica, não relacionada à doença, ou até mesmo uma manifestação de apresentação da mesma(1-6).
Ceratoconjuntivite seca é descrita em 25% dos pacientes com artrite reumatóide, esclerite é a segunda manifestação ocular mais freqüente ocorrendo em 0,67% a 6,3% dos casos, seguida pela ceratite que mais comumente se desenvolve em contigüidade com esclerite, ocorrendo em 37 a 50% dos pacientes com esclerite embora também possa ocorrer como um achado isolado(1,2,4).
O aparecimento de esclerite necrosante ou ceratite ulcerativa periférica no curso clínico da artrite reumatóide pode refletir a presença de lesões vasculíticas viscerais e estão relacionadas com um prognóstico reservado(1-8). Portanto, é importante que os oftalmologistas estejam familiarizados com este grupo de doenças, porque podem causar alta morbidade ocular e sistêmica se incorretamente diagnosticadas ou tratadas e estas geralmente requerem terapia sistêmica para melhora do prognóstico ocular e sistêmico da doença.
Neste trabalho, nós descrevemos nove casos de pacientes portadores de doenças do tecido conectivo que apresentaram envolvimento do segmento anterior, com ênfase nas características clínicas, doença sistêmica associada, resposta ao tratamento e evolução.
MÉTODO
Descrevemos as manifestações oculares de segmento anterior em uma série de nove pacientes com doença do tecido conectivo, nos quais o acometimento ocular foi a primeira manifestação da doença sistêmica ou ocorreu durante o curso da doença já previamente diagnosticada. Os pacientes foram acompanhados prospectivamente com relação às manifestações oculares, diagnóstico, resposta ao tratamento e evolução.
Todos os pacientes foram atendidos no setor de Doenças Externas Oculares e Córnea da Universidade Federal de São Paulo (UNIFESP) e encontravam-se em tratamento ambulatorial no período deste estudo (julho/1999 - dezembro/2000).
Os pacientes portadores de doença ocular de natureza inflamatória autoimune que são atendidos no nosso ambulatório são submetidos a exame oftalmológico completo que inclui anamnese detalhada, pesquisa dos antecedentes oculares e da história patológica pregressa, medida da acuidade visual, tonometria de aplanação, biomicroscopia e oftalmoscopia binocular indireta. Para a investigação de ceratoconjuntivite seca avaliou-se a estabilidade do filme lacrimal através da medida do tempo de quebra do filme lacrimal, considerando-se valor menor ou igual a 10 segundos anormal. Realizou-se também o teste da rosa bengala, considerando-se como critério diagnóstico a classificação proposta por van Bijsterveld (1969), baseada na intensidade e localização da coloração utilizando uma escala de 0 a 3 em três áreas: conjuntiva nasal, conjuntiva temporal e a córnea, considerando-se anormal pontuação igual ou maior que 3 em um olho e o teste de Schir-mer I para avaliação quantitativa da produção basal e reflexa da lágrima, considerando-se sugestivo de olho seco valores menores ou iguais a 10 milímetros de umidade no papel de filtro em 5 minutos. Considerou-se como diagnóstico a presença de sintomas e sinais de olho seco (sensação de areia nos olhos, queimação, hiperemia, diminuição do menisco lacrimal, presença de muco no fundo se saco conjuntival e de crostas nos cílios) associado a tempo de quebra do filme lacrimal anormal com ou sem anormalidades nos testes de Schirmer e rosa bengala.
Todos os pacientes foram submetidos à avaliação clínica com revisão dos sistemas respiratório, circulatório, ósteoarticular, gastrointestinal, dermatológico, além da investigação laboratorial que incluiu hemograma, velocidade de hemossedimentação, glicemia, uréia, creatinina, sorologia para sífilis, reação de Mantoux, exame de urina tipo I, e provas imunológicas (fator reumatóide, anticorpos antinucleares, crioglobulinas, complemento e anticorpos anticitoplasma de neutrófilo). Além da realização de radiografia de tórax e de seios da face.
O diagnóstico da doença sistêmica foi realizado segundo os critérios do Colégio Americano de Reumatologia para o diagnóstico de artrite reumatóide (1988), da esclerose sistêmica (1980) e da granulomatose de Wegener (1990)(10).
O diagnóstico das esclerites baseou-se nos achados oculares de olho vermelho com congestão dos vasos episclerais profundos, edema escleral, presença de nódulos ou ainda áreas de necrose ou de afilamento escleral. Utilizou-se a classificação proposta por Watson e Hayreh (1976) que dividem as esclerites anteriores em difusa, nodular e necrosante com e sem inflamação, sendo esta última também conhecida como escleromalacia perforans(3-4,6,8). Nos casos com presença de infiltrados e/ou ceratite periférica adjacente à esclerite, procedeu-se ao recuo e à ressecção da conjuntiva do limbo para minimizar a ação de células inflamatórias e de mediadores da inflamação que chegam à periferia da córnea através dos vasos do limbo e que podem levar à destruição do estroma da córnea.
A conduta adotada no setor para o tratamento inicial de pacientes com esclerite anterior difusa ou nodular associada a doenças do tecido conectivo consiste no uso de antiinflamatório não hormonal sistêmico, o qual é mantido por tempo prolongado (6 meses ou mais) e corticóide tópico. Nos casos que evoluem sem melhora do quadro ocular ou que apresentem recorrência da doença na vigência do tratamento utilizamos, como segunda opção, corticóide sistêmico, que é usado por curto período (2 semanas) com rápida descontinuação da medicação. Se não houver melhora da esclerite e/ou ceratite, a terapêutica é substituída ou acrescida de um imunossupressor. Nas esclerites nodulares, unilaterais, não acompanhadas de envolvimento corneal, o metotrexate (7,5 a 15 mg/semana) tem sido utilizado com bons resultados, alternativamente também temos utilizado com sucesso a ciclosporina A (3,0-5,0 mg/kg/dia). Nas esclerites necrosantes, rapidamente progressivas e destrutivas, com ou sem envolvimento da periferia da córnea, bem como nas esclerites de qualquer tipo associadas a granulomatose de Wegener, a ciclofosfamida (15,0 mg/kg em pulso endovenoso ou dose oral de 2,0 mg/kg/dia) tem sido o imunossupressor de primeira escolha. A avaliação sistêmica, bem como a orientação e a monitorização da terapêutica foram realizados em colaboração com a disciplina de reumatologia da UNIFESP.
A intervenção cirúrgica é realizada nos casos que evoluem com destruição tecidual com risco de perfuração ocular. Os procedimentos utilizados foram a aplicação de adesivo tecidual de cianoacrilato e adaptação de lente de contato terapêutica nas ulcerações corneais de até três milímetros de diâmetro, com cobertura antibiótica profilática com colírio de tobramicina quatro vezes ao dia, como medida provisória na tentativa de manter a integridade do bulbo ocular. Nas lesões maiores envolvendo a córnea ou a esclera realizou-se transplante tectônico de córnea ou enxerto de esclera, respectivamente.
RESULTADOS
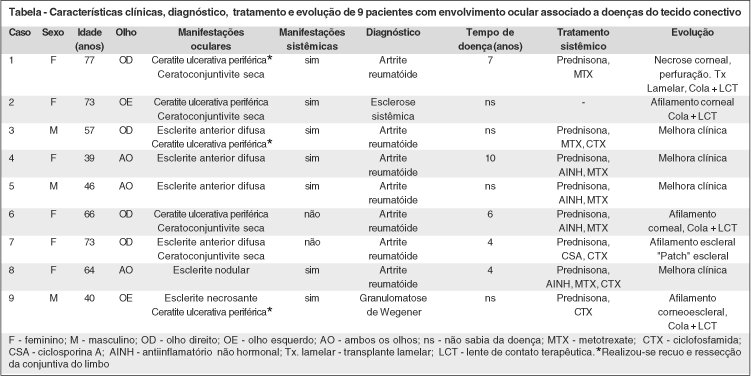
Foram atendidos nove pacientes com quadro de esclerite e/ou ceratite ulcerativa periférica secundários a doenças do tecido conectivo no ambulatório de doenças externas oculares e córnea no período deste estudo (julho/1999-dezembro/2000). Destes pacientes, cinco (56%) eram sabidamente portadores de artrite reumatóide e em quatro pacientes (44%) o diagnóstico da doença subjacente foi realizado a partir da avaliação clínica e da investigação laboratorial, iniciados após o aparecimento da lesão ocular que evidenciaram que dois pacientes eram portadores de artrite reumatóide (casos 3 e 5), um de esclerose sistêmica (caso 2) e outro de granulomatose de Wegener (caso 9). Todos os casos são apresentados na tabela.
A média de idade dos pacientes foi de 59,4 anos (variando de 39 a 77 anos), sendo que 66,7% eram do sexo feminino e 33,3% do sexo masculino. O envolvimento ocular foi unilateral em 67% dos casos, acometendo predominantemente OD (2:1).
As manifestações oculares mais freqüentes foram esclerite em 66,6% (6 casos), seguida por ceratite ulcerativa periférica em 55,5% (5 casos) e ceratoconjuntivite seca em 44,4% (4 casos).
Oitenta e três por cento dos pacientes com esclerite (5 casos) eram portadores de artrite reumatóide, destes, quatro apresentaram a forma anterior difusa (casos 3, 4, 5 e 7) e um, a forma nodular (caso 8). O paciente que apresentou esclerite necrosante teve o diagnóstico de granulomatose de Wegener (caso 9). Do total de pacientes que apresentaram ceratite ulcerativa periférica (5 casos), três eram portadores de artrite reumatóide (casos 1, 3 e 6), um de esclerose sistêmica (caso 3) e o outro de granulomatose de Wegener (caso 9). Dois pacientes apresentaram ceratite ulcerativa periférica e esclerite concomitantemente, sendo estes portadores de artrite reumatóide (caso 3) e granulomatose de Wegener (caso 9). Setenta e cinco por cento dos pacientes que apresentavam ceratoconjuntivite seca eram portadores de artrite reumatóide (casos 1, 6 e 7) e um teve o diagnóstico de esclerose sistêmica (caso 2).
Corticóide sistêmico (prednisona, na dose de 1mg/kg/dia) foi a medicação prescrita no tratamento inicial das inflamações oculares em 89% dos pacientes. Entretanto, não controlou a inflamação ocular em 62% dos casos e 38% apresentaram intolerância à medicação (distúrbios gastrointestinais) ou ainda eram portadores de diabetes mellitus ou hipertensão arterial. Falha da terapêutica com corticóide sistêmico foi considerada após um período médio de duas semanas de uso da medicação, sem melhora da doença ocular. Nestes casos, iniciamos os agentes citotóxicos (metotrexate/ ciclofosfamida) e ainda a ciclosporina A. A maioria (89%) dos pacientes requereu terapia imunossupressora para o controle do processo inflamatório ocular. O corticosteróide oral foi mantido em baixas doses (5-10 mg/dia) combinados com a terapia imunossupressora nos casos que apresentavam esclerite nodular ou necrosante ou que apresentassem destruição da periferia da córnea ou, ainda, naqueles que apresentavam alguma indicação sistêmica para uso dos mesmos. Cinqüenta e cinco por cento dos pacientes usavam corticosteróide oral, em baixas doses (5-10 mg/dia), quando foram referidos ao ambulatório de oftalmologia (casos 3, 4, 5, 6 e 7).
O metotrexate (7,5-15 mg/semana) foi o agente citotóxico de primeira escolha em 75% dos casos. Nas inflamações que evoluem com necrose tecidual, de rápida progressão, ou naquelas em que não obtivemos controle da doença com metotrexate (37,5%), iniciamos ciclofosfamida na forma de pulsos endovenosos de 15 mg/kg a cada 3-4 semanas (3-4 pulsos) ou dose oral de 2 mg/kg/dia, a qual foi mantida por aproximadamente 6 meses. No paciente com granulomatose de Wegener ocular realizamos pulsoterapia de metilprednisolona (1000 mg/dose, endovenoso - 3 pulsos), seguida por ciclofosfamida oral (2 mg/kg/dia), observando-se estabilização do quadro na primeira semana de tratamento e em uma paciente (caso 7), antes de iniciarmos a ciclofosfamida, utilizamos a ciclosporina A durante 45 dias, como droga de segunda escolha, entretanto, sem melhora da inflamação ocular.
Neste grupo de pacientes não tivemos efeitos colaterais atribuídos aos imunossupressores.
Tratamento cirúrgico foi realizado em todos os casos com ceratite ulcerativa periférica e/ou esclerite necrosante (casos 1, 2, 3, 6, 9). Nestes casos, os procedimentos foram realizados concomitantemente com o tratamento sistêmico da doença de base e incluíram recuo e ressecção da conjuntiva (60%), aplicação de tecido adesivo de cianoacrilato e adaptação de lente de contato terapêutica (80%) e transplante tectônico de córnea em 20% dos casos. Duas pacientes (casos 2 e 6), apresentaram ceratite ulcerativa periférica a qual foi relacionada a ceratoconjuntivite seca, tendo sido realizado a aplicação de adesivo tecidual. Na paciente com esclerose sistêmica (caso 2) não introduzimos terapia sistêmica pela ausência de manifestações de atividade sistêmica da doença. Uma paciente (caso 1), referida ao ambulatório de oftalmologia com quadro de úlcera de córnea e perfuração ocular, foi submetida a transplante tectônico de córnea, evoluindo no pós-operatório com necrose do enxerto, tendo sido tratada com aplicação local de tecido adesivo.
Em outra paciente (caso 7), que evoluiu com afilamento escleral e exposição do tecido uveal, realizou-se enxerto de esclera após melhora da inflamação ocular.
Remissão da doença ocular foi obtida em um período médio de três meses, após o início da terapia imunossupressora, sem se observar recorrências durante o período de seguimento (julho/99 - dezembro/00).
DISCUSSÃO
Entre as doenças difusas do tecido conjuntivo, a artrite reumatóide é a mais comum, afetando cerca de 1% da população geral e, destes, aproximadamente 25% irão apresentar complicações oculares em algum momento durante o curso da doença(1-5,10).
Na nossa série, 77,8% dos pacientes eram portadores de artrite reumatóide. A distribuição, quanto ao sexo dos nossos pacientes, refletiu a maior incidência da doença no sexo feminino (razão mulher/homem 5:2).
Diferentemente da literatura, a manifestação ocular mais comum na nossa série foi a esclerite em 66,6% dos casos, seguida por ceratite ulcerativa periférica (55,5%) e ceratoconjuntivite seca (44,4%). É importante enfatizar que estes achados não traduzem a realidade de nossa população de pacientes com artrite reumatóide, uma vez que selecionamos apenas os casos que procuraram o nosso ambulatório por apresentarem comprometimento ocular grave, requerendo imunossupressão sistêmica.
Esclerite geralmente ocorre em pacientes com artrite reumatóide bem estabelecida, que têm outras manifestações articulares e/ou extra-articulares da doença(2-8). É com freqüência um sinal de piora da doença sistêmica e razão para reavaliação da terapêutica destes pacientes(2-8). Todos os tipos de esclerite anterior, incluindo a difusa, nodular, necrosante com inflamação e necrosante sem inflamação (escleromalacia perforans), bem como a esclerite posterior, têm sido documentados em pacientes com artrite reumatóide(2-4,6,8). Ceratite ulcerativa periférica mais comumente se desenvolve em contigüidade com uma esclerite, ocorrendo em 37 a 50% dos pacientes com esclerite, mas também pode ocorrer como um achado isolado(1-5). O aparecimento de esclerite necrosante com inflamação e ceratite ulcerativa periférica deve ser considerado uma emergência oftalmológica e médica(3,7). Este grupo de pacientes tem um prognóstico reservado devido à elevada associação com lesões vasculíticas sistêmicas potencialmente letais(3,6-7). Mais importante, sem terapia imunossupressora sistêmica apropriada, 50 a 60% destes pacientes morrem em 5 anos do início da esclerite(7).
Além da artrite reumatóide, um paciente apresentou esclerite necrosante e ceratite ulcerativa periférica, como única manifestação clínica de granulomatose de Wegener e outro paciente apresentou ceratoconjuntivite seca e ceratite ulcerativa, como parte do quadro de esclerose sistêmica. Em ambos os casos, o diagnóstico da doença foi realizado a partir da investigação clínica e laboratorial iniciadas após o aparecimento da lesão ocular.
Assim, considerando-se a gravidade destes quadros e as complicações locais e sistêmicas associadas, é fundamental o reconhecimento precoce destas doenças e a instituição da terapia apropriada(1-8).
O tratamento clínico recomendado para as inflamações oculares associadas à artrite reumatóide inclui o uso de antiinflamatórios não hormonais, corticosteróide e quimioterapia imunossupressora sistêmica(3-8,11-14).
Neste grupo de pacientes o uso isolado de corticosteróide sistêmico não permitiu o controle do processo inflamatório ocular em 55% dos casos. Segundo a literatura, o uso de corticosteróide sistêmico é freqüentemente incapaz de interromper a inflamação ocular progressiva e destrutiva associada à artrite reumatóide(3-8). Pulsoterapia intravenosa com doses imunossupressoras de metilprednisolona foi reportada ser eficaz em pacientes que não responderam a corticosteróide oral(11). Entretanto, o segmento a longo prazo destes pacientes é desconhecido, e devemos sempre considerar a possibilidade do aparecimento de efeitos colaterais da terapia esteróide em altas doses por tempo prolongado(11). Além disto, corticosteróide sistêmico não influencia na elevada taxa de mortalidade de pacientes com vasculíte reumatóide(7), como também não melhora o prognóstico de pacientes com granulomatose de Wegener(15).
Terapia imunossupressora tem sido usada nas inflamações oculares associadas à artrite reumatóide(2-8,11-14), e está certamente indicada em condições letais como vasculites reumatóides, granulomatose de Wegener e outras.
A ciclofosfamida (CTX) foi o agente mais eficaz no nosso grupo de estudo. O metotrexate (MTX), uma droga com menos toxicidade potencial, quando usada uma vez por semana nas dosagens recomendadas(3,8,11-13), também provou ser altamente benéfica. O metotrexate é considerado uma boa opção no tratamento inicial da ceratite ulcerativa periférica e esclerite, na maioria dos pacientes com artrite reumatóide, reservando agentes com toxicidade potencialmente maior, como a ciclosfosfamida(3,8,11-13), para falha terapêutica, intolerância a droga, e para casos nos quais a destruição ocular é rapidamente progressiva e destrutiva. A ciclosporirina A e azatioprina também são boas opções terapêuticas e têm sido usadas como drogas de segunda escolha(3,5,8,11-13).
Ciclofosfamida é a droga de escolha no tratamento da granulomatose de Wegener, independentemente da forma de esclerite que o paciente apresente. Segundo a literatura, não tratar estes pacientes desta forma representa negligência, diante da mortalidade associada com terapias alternativas(1,3-4,6,8,15). Corticosteróide é útil como terapia adjuvante(1,3,6,8,15).
O tratamento cirúrgico é também um importante componente no tratamento de pacientes com destruição da córnea e da esclera associada a doenças auto-imunes (3,5,16-19). Os procedimentos por nós utilizados foram ressecção conjuntival, adesivo de cianoacrilato e transplante lamelar tectônico.
A ressecção conjuntival parece interromper, temporariamente, os eventos imunológicos locais que levam à ulceração corneal. Entretanto, a experiência publicada na literatura internacional, atesta que a cirurgia isoladamente é incapaz de influenciar o processo imunológico subjacente, e que é insuficiente no tratamento das ceratites ulcerativas periféricas, associadas a doenças auto-imunes(3,5,16).
Ceratoplastia lamelar ou penetrante pode ser indicada em pacientes com ulceração corneal para prevenir perfuração de úlceras muito grandes para uso de tecido adesivo ou para reabilitação visual (3,5,17-19). Entretanto, a ceratoplastia tem um prognóstico ruim nestes casos, por causa da probabilidade de posterior destruição do enxerto pela doença de base, pela formação de defeito epitelial e ulceração estromal, pela ceratoconjuntivite seca, e hipestesia(3,5,18-19).
O envolvimento da córnea e da esclera é, com freqüência, uma manifestação de gravidade das doenças do tecido conectivo, devendo pois, os oftalmologistas estarem alertas para a sua detecção precoce, sendo o tratamento com drogas imunossupressoras eficaz no controle do envolvimento ocular.
REFERÊNCIAS
1. Shiuey Y, Foster CS. Peripheral Ulcerative Keratitis and Collagen Vascular Disease. Int Ophthalmol Clin. 1998;38(1):21-32.
2. Harper SL, Foster, CS. The Ocular Manifestations of Rheumatoid Disease. Int Ophthalmol Clin. 1998;38(1):1-19.
3. Foster CS, Maza MS M. The Sclera. NewYork (NY): by Springer-Verlag;1994.
4. Hamideh F, Prete PE. Ophthalmologic Manifestations of Rheumatic Diseases. Semin Arthritis Rheum. 2001;30(4):217-41.
5. Messmer EM, Foster CS. Destructive Corneal and Scleral Disease Associated with Rheumatoid Arthritis. Cornea. 1995;14(4):408-17.
6. Maza MS, Foster CS, Jabbur NS. Scleritis Associated with Systemic Vasculitic Diseases. Ophthalmology. 1995;102(4):687-92.
7. Foster CS, Forstot SL, Wilson LA. Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis. Ophthalmology. 1984;91(10):1253-63.
8. Watson PA. The diagnosis and management of scleritis. Ophthalmology. 1980;87(7):716-20.
9. Van Bijsterveld OP. Diagnostic tests in the Sicca Syndrome. Arch Ophthalmol. 1969;82(1):10-4.
10. Moreira C, Carvalho MAP. Reumatologia Diagnóstico e Tratamento. São Paulo: Editora Médica e Científica Ltda; 2001.
11. Meyer PAR, Watson PG, Franks W, Dubord P. Pulsed immunosuppressive therapy in the treatment of immunologically induced corneal and scleral disease. Eye. 1987;1(Pt 4):487-95.
12. Jampol LM, West C, Goldberg MF. Therapy of scleritis with cytotoxic agents. Am J Ophthalmol. 1978;86(2):266-71.
13. Hemady R, Tauber J, Foster CS. Immunosuppressive Drugs in Immune and Inflammatory Ocular Disease. Surv Ophthalmol. 1991;35(5):369-85.
14. Clements PJ, Davis J. Cytotoxic drugs: their clinical application to the rheumatic diseases. Semin Arthritis Rheum.1986;15(4):231-54.
15. Haynes BF, Fishman ML, Fauci AS, Wolff SM. The ocular manifestations of Wagener's granulomatosis. Fifteen years experience and review of literature. Am J Med. 1977;63(1):131-41.
16. Feder RS, Krachmer JH. Conjunctival resection for the treatment of the rheumatoid corneal ulceration. Ophthalmology. 1984;91(2):111-5.
17. Webster RG Jr, Slansky HH, Refojo MF, Boruchoff SA, Dohlman CH. The use of adhesive for the closure of corneal perforations. Report of two cases. Arch Ophthalmol. 1968;80(6):705-9.
18. Palay DA, Stulting RD, Waring GO 3rd, Wilson LA. Penetrating keratoplasty in patients with rheumatoid arthritis. Ophthalmology. 1992;99(4):622-7.
19. Bernauer W, Ficker LA, Watson PG, Dart JKG. The Management of Corneal Perforations Associated with Rheumatoid Arthritis. Ophthalmology. 1995; 102(9):1325-37.
Endereço para correspondência
Rua Prof. Clemente Fortes, 2390
Teresina (PI) CEP 64051 - 030
E-mail: [email protected]
Recebido para publicação em 03.06.2002
Versão revisada recebida em 27.02.2004
Aprovação em 12.04.2004

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}