Arq. Bras. Oftalmol. 2004; 67 (4): 10.1590/S0004-27492004000400012
Total: 1975
Mário Teruo Sato1; Rodrigo Marzagão2; Nina Amália Brancia Pagnan3; Newton Freire-Maia3; Carlos Augusto Moreira Júnior2
DOI: 10.1590/S0004-27492004000400012
RESUMO
OBJETIVOS: Propor complementação da atual classificação do eletrorretinograma (ERG) com a inclusão deste novo tipo de onda, discutir os possíveis mecanismos para a distrofia retiniana com onda rápida escotópica (DRORE) associada à síndrome dos cabelos anágenos frouxos (SCAF) e análise do heredograma da família estudada. MÉTODO: Foram descritos na parte I deste trabalho. RESULTADOS: A análise do heredograma da família demonstrou tratar-se de herança autossômica recessiva com expressão parcial no heterozigoto; outros resultados foram descritos na parte I deste trabalho. CONCLUSÕES: Por se tratar do primeiro relato na literatura, os achados descritos sugerem fortemente que a distrofia retiniana com onda rápida escotópica associada à síndrome dos cabelos anágenos frouxos pode ser uma nova entidade nosológica. Neste trabalho propomos uma classificação inédita de todas as distrofias maculares e degenerações retinianas associadas a distúrbios capilares no grupo B das displasias ectodérmicas.
Descritores: Eletrorretinografia; Degeneração retiniana; Displasia ectodérmica; Hipotricose; Alopecia; Epitélio pigmentado ocular
ABSTRACT
PURPOSE: To propose the complementation of present classification of the electroretinogram (ERG) with the inclusion of this new kind of wave, to discuss the possible mechanisms for the scotopic fast wave retinal dystrophy (SFWRD) associated with the loose anagen hair syndrome (LAHS) and to analyze the pedigree of the studied family. METHODS: Were described in part I of this study. RESULTS: The pedigree analysis of the family showed to be an autosomal recessive form of inheritance with partial expression in the heterozygote; other results were described in part I of this study. CONCLUSION: Being the first account in the literature, the described finding strongly suggests that scotopic fast wave retinal dystrophy associated with the loose anagen hair syndrome can be a new nosological entity. In this study, we propose an unpublished classification of all macular dystrophies and retinal degenerations associated with hair disorders in group B of ectodermal dysplasias.
Keywords: Electroretinography; Retinal degeneration; Ectodermal dysplasia; Hypotrichosis; Alopecia; Pigment epithelium of eye
INTRODUÇÃO
Nesta segunda parte do trabalho discutiremos os aspectos de classificação deste novo tipo de onda do eletrorretinograma (amplitude e tempo de culminação diminuídos), possíveis mecanismos para o comportamento paradoxal desta, a hereditariedade e uma classificação envolvendo todas as distrofias maculares e degenerações retinianas descritas associadas a distúrbios capilares no grupo B das displasias ectodérmicas.
MÉTODOS
Foram descritas na parte I deste trabalho(1).
RESULTADOS
A análise genealógica da família (Figura 1 e Tabela 1)(1) sugere fenótipo com herança autossômica recessiva e expressão parcial no heterozigoto.
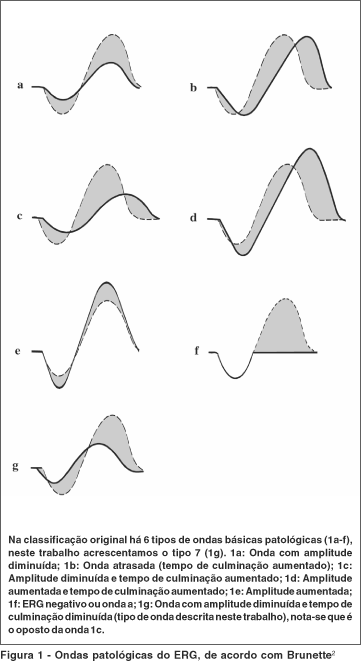
DISCUSSÃO
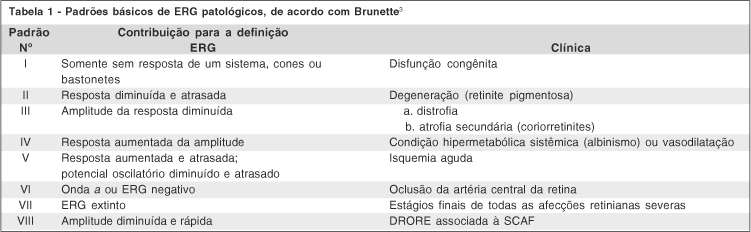
Na primeira parte do trabalho descrevemos um novo tipo de onda do eletrorretinograma (ERG) que deu nome a esta distrofia macular. Sugerimos então, uma complementação da classificação de Brunette das alterações das ondas do ERG, incluindo esta onda caracterizada por resposta rápida e amplitude diminuída. A distrofia retiniana com onda rápida escotópica (DRORE) associada à síndrome dos cabelos anágenos frouxos (SCAF) pode ser incluída no sétimo tipo de ondas patológicas do ERG(2) (figura 1), ou no padrão VIII da classificação dos padrões básicos de ERG patológicos(3) (tabela 1).
Como hipóteses prováveis, dois mecanismos são possíveis para o encurtamento do tempo de culminação das ondas da fase escotópica como se explicita a seguir.
1) Distúrbio molecular e genético: Mutações que acarretem a ausência ou a produção de formas anômalas de moléculas que atuem no metabolismo normal da retina podem estar envolvidas da determinação desse fenótipo (DRORE e SCAF). Sinais intercelulares anormais e distúrbios das moléculas de adesão dos folículos, como as desmogleínas e as caderinas. Essas moléculas se encontram ausentes entre as camadas nas quais previamente a bainha radicular interna estava aderida na SCAF, encontrando-se desintegrada e cornificada(4). Possivelmente pode haver um gene que codifique essas moléculas de adesão dos folículos associados ao problema na retina, de fato foi identificado uma mutação no CDH3, gene que codifica a molécula P-caderina, proteína expressa no epitélio pigmentado da retina (EPR) e nos folículos pilosos em pacientes com distrofia macular e hipotricose congênita(5).
2) Ação de agentes virais, tóxicos ou imunológicos: O eletro-oculograma (EOG) mostrou-se alterado em pacientes com alopécia areata e sem alterações oculares, sugerindo alteração no EPR e nos cabelos, por ação de insultos virais, tóxicos ou imunológicos(6).
3) Hipótese eletroneurofisiológica: As alterações que ocorrem na retina para a formação da onda a e b do ERG, são através da variação do potencial de membrana celular, que ocorrem nos fotorreceptores (1/3 externo da retina), responsável pela onda a e interação das células bipolares e as de Muller que formam a onda b (1/3 médio da retina), o potencial de ação é formado a partir das células ganglionares que formam o nervo óptico (1/3 interno da retina). A onda a é formada pelo fechamento dos canais de Na (sódio) nos fotorreceptores, na corrente escura e com conseqüente hiperpolarização da célula, a onda b é formada pela despolarização das células bipolares e pelo aumento do K (potássio) extracelular nas células de Muller(7). Duas hipóteses são aventadas: 1) Mutação na região helicoidal S4 do canal de Na, local em que se acredita estar o sensor de voltagem e que é uma estrutura altamente sensível à mudança no campo elétrico através da membrana, já que foi demonstrado em experimento, por mutação neste local, com a posterior redução da carga positiva em S4 que abaixa a sensitividade da ativação do canal de voltagem do Na(8), o oposto com o aumento da carga positiva em S4 e aumento da sensitividade de ativação do canal de Na possam ocorrer na DRORE, o que aumentaria a velocidade da formação do potencial de membrana. 2) A proteína G ou transducina tem 3 subunidades a, ß, e g (Taßg. GDP). A subunidade g está fixada ao GDP, nos fotorreceptores quando a rodopsina é ativada pela luz causa a subunidade Ta trocar a sua ligação de GDP por GTP, que causa o seu desligamento da subunidade Tßg. O complexo GTP e Ta é que produz os efeitos biológicos com a ativação da fosfodiesterase, que hidrolisa o cGMP e ocorre o fechamento dos canais de Na e a hiperpolarização da célula, que originam a onda a do ERG. A atividade da GTPase intrínseca da subunidade Ta converte o GTP em GDP. Então uma mutação em Ta poderia reduzir a atividade da GTPase intrínseca e reduzir a conversão de GTP a GDP com isto os fotorreceptores ficariam mais ativos, por ficar só uma via de GDP para GTP o que aumentaria a velocidade da fototransdução, que seria traduzido na DRORE com a onda mais rápida, esta hipótese é baseado em um dos exemplos de mutação da proteína G, responsável pela regulação dos fatores do crescimento, que é a subunidade Gsa que reduz a atividade intrínseca da GTPase, como resultado ele fica mais ativa nos somatotropos da hipófise, desenvolvendo níveis permanentemente aumentados de AMP cíclico. Isso faz com que essas células se tornem neoplásicas, produzindo adenomas hipofisários que causam acromegalia(9). Outros pontos de mutações possíveis para se aumentar a velocidade da fototransdução seria na rodopsina ativada e na fosfodiesterase que são os locais em que uma amplificação da resposta a cascata do cGMP acontece(10). Estes eventos descritos provocariam a hiperpolarização dos fotorreceptores, com conseqüente redução na liberação de neurotransmissores nas sinapses, que é a origem da onda a do ERG, o que ocasiona a despolarização das células bipolares, provocando um aumento do K extracelular, causando a despolarização das células de Muller, gerando assim, a onda b do ERG. Todos estes eventos aconteceriam mais rapidamente que o normal nos pacientes com a DRORE, com um encadeamento de eventos, através da hiperpolarização rápida dos fotorreceptores (formação rápida da onda a), que despolarização precocemente as células bipolares, que por conseqüência aumentaria rapidamente o K extracelular, havendo a despolarização rápida da célula de Muller, deflagrando, assim precocemente a onda b do ERG.
Na família estudada, observamos quatro afetados (dois homens e duas mulheres) numa irmandade de cinco pessoas, cujos pais não apresentavam a síndrome. Além disso, os pais dos afetados nasceram na mesma cidade pequena (Morretes), e a mãe (caso II-19) informou-nos sobre a existência de consangüinidade remota de grau ignorado entre ela e seu marido (caso II-20). Esse fato, além das evidências do heredograma na figura 1(1), apóia a hipótese de herança autossômica recessiva para o fenótipo estudado.
A observação ao microscópio óptico e eletrônico de alterações capilares na mãe (tabela 1, caso II-19)(1) e no irmão sem DRORE (caso III-20), numa intensidade menor que as evidenciadas nos indivíduos com a DRORE, poderia ser interpretada como evidência de manifestação no heterozigoto. De fato, manifestações mais brandas de distúrbios autossômicos recessivos podem ocasionalmente ser expressas em heterozigotos e já foram relatadas(11), por exemplo, descreveram um paciente com síndrome de Tel-Hashomer (também condicionada por gene autossômico recessivo) e procederam à revisão da literatura. Essa síndrome caracteriza-se pela ocorrência de sinais faciais (hipertelorismo ocular e filtro nasal longo), camptodactilia, pés tortos, hipoplasia muscular e alterações dermatoglíficas. Os autores admitem que a observação de camptodactilia e de alterações dermatoglíficas em pais de afetados pode ser interpretada como evidência de manifestação no heterozigoto.
O caso da DRORE associada à SCAF, que descrevemos pela primeira vez neste trabalho, requer considerações semelhantes. Embora a existência de consangüinidade entre os pais apóie a hipótese de herança autossômica recessiva, não se pode deixar de considerar a hipótese de tratar-se de fenótipo determinado por gene autossômico dominante com expressividade muito variável. Nesse caso, a manifestação de sinais na mãe e num dos irmãos seria interpretada como evidência da extrema variabilidade fenotípica. Acreditamos, portanto, existirem elementos suficientes que permitam supor que, na família estudada neste trabalho, os indivíduos (caso II-19, III-20), com sinais mais brandos, possam ser considerados como heterozigotos quanto ao gene que determina o quadro clínico desta entidade. Neste trabalho, os resultados (Tabela 1)(1) sugerem fortes evidências da relação direta entre cabelos e olhos, já que os pacientes (casos II-19 e III-20) apresentaram sinais da SCAF (na MO e MEV), sem, no entanto, apresentarem clínica de SCAF (pacientes negavam fios de cabelos facilmente arrancáveis quando puxados e referiam que os cabelos cresciam normalmente) e sem apresentarem acometimento oculares (DRORE), sendo considerados como prováveis heterozigotos. Ao contrário, os pacientes que apresentaram clínica da SCAF apresentaram também problemas oculares (casos III-21 a III-24), ou seja, o problema ocular (DRORE) parece estar relacionado diretamente com a gravidade do distúrbio capilar, sendo estes pacientes considerados como prováveis homozigotos. As evidências apresentadas anteriormente descartam que a associação descrita neste trabalho seja de fatos coincidentes entre a SCAF e distrofia macular (DRORE).
Uma possível explicação das diferenças observadas na expressão da distrofia, entre os irmãos e irmãs afetados, com a presença de coloboma macular nas irmãs, poderia ser pela ação de genes modificadores que influenciariam a manifestação fenotípica, durante o desenvolvimento. Esses estudos trazem a perspectiva de elucidação dos mecanismos que levam à variabilidade fenotípica, como demonstram, por exemplo, os resultados obtidos que sugeriram a existência de um gene modificador através de análise de ligação numa família afetada pela síndrome de Van der Woude (autossômica dominante com expressividade variável e penetrância incompleta)(12). Estas observações apóiam a hipótese de um gene no braço curto do cromossomo 17, que, quando presente juntamente com o gene da síndrome (cromossomo 1, braço curto), aumenta a probabilidade de que o indivíduo tenha fissura de palato(12).
O esclarecimento quanto ao modo de herança, possível heterogeneidade, delineamento fenotípico, identificação do gene responsável e suas mutações, bem como do produto protéico e suas funções só serão possíveis com a descrição de novas famílias. Isso possibilitará que o gene determinante da DRORE associada à SCAF seja identificado e clonado, permitindo realizar estudos através da análise direta do DNA. Amostras de sangue periférico, com a extração do DNA, foram coletadas dos pacientes examinados, visando a estudos posteriores que poderão contribuir para o conhecimento da localização do gene. A localização do gene provavelmente seja importante para se elucidar o que determina o curso do tempo (time course) de uma onda, que é representado no ERG pelo tempo de culminação, através das pesquisas dos prováveis locais de mutações descritos anteriormente.
Há somente um relato da associação entre a SCAF e alterações oculares (coloboma ocular)(13). Não se encontram relatos na literatura mundial, até o presente momento, entre a distrofia macular e a SCAF. Em face dos achados incomuns, encontrados na descrição desta família, somos levados a considerar que essas informações evidenciam o fato de que podemos estar diante de nova entidade nosológica, dentro do grupo B das displasias ectodérmicas. Sendo assim, uma nova classificação foi sugerida, incluindo a associação descrita neste trabalho dentro do grupo B das displasias ectodérmicas(14-15). Etimologicamente, o termo displasia significa crescimento anormal, que é diferente de disgenesia (geração anormal)(15). O termo distrofia etimologicamente é restrito a condições que se desenvolvem algum tempo após o nascimento, tais como as distrofia miotônica ou distrofias progressivas musculares(15). Displasia ectodérmica caracteriza-se essencialmente por três condições: 1) a maioria dos distúrbios afeta os tecidos de origem ectodérmica; 2) estes distúrbios são de desenvolvimento e 3) a hereditariedade tem um papel causal importante. Freire-Maia apresentou uma definição clínica e uma classificação clínico-mnemônica das displasias ectodérmicas(14). Essa classificação é artificial e foi formulada somente com o intento de delimitar o campo. Afecções que afetam somente a pele e a mucosa oral (ictiose congênita), que têm sido já classificadas como displasia ectodérmica por Greene GW apud(16), não serão cobertas por essa definição(14-15). As displasias ectodérmicas nem sempre se caracterizam por sinais de origem exclusivamente ectodérmicas, podendo conter também sinais de múltiplas origens embriológicas. O nome original permanece somente por razões históricas e porque o defeito ectodérmico é mais grave ou mais aparente que os outros(15). As displasias ectodérmicas são divididas em dois grupos: O grupo A contém displasias com pelo menos dois dos quatro sinais: 1-tricodisplasia (cabelos), 2 - defeitos dentários (dentes), 3 - oni-codisplasia (unhas) e 4- disidrose (glândulas sudoríparas). Os diferentes subgrupos podem ser chamados pela combinação dos números acima citados ou pela combinação da palavra de origem grega, referindo a estrutura que mostra o distúrbio. Como exemplo, temos: 1-2-3-4, que é igual à trico-odonto-ônico-disidrótico. Afecções com pelo menos um dos quatro sinais citados acima mais pelo menos um sinal de outra estrutura de origem ectodérmica (tarjado 5) são classificadas dentro de outro grupo (grupo B). Essas afecções podem ser classificadas pela combinação dos respectivos números ou pela palavra de origem grega, referindo-se a um dos quatro sinais básicos. Como exemplo, temos: pêlo torto e surdez pertencem ao subgrupo 1-5 ou subgrupo tríquico. O grupo B parece ser bem menor que o grupo A(15).
Devido ao fato de haver várias descrições envolvendo cabelo e olhos na literatura médica, ou seja, distúrbios dos cabelos e distrofias ou degenerações oculares, propomos uma classificação dessas alterações no grupo B das displasias ectodérmicas. A associação entre cabelo e olho, como parte de um distúrbio dentro das displasias ectodérmicas, não é nova(17-19); mas estes autores não sugeriram nenhuma classificação como grupo dentro das displasias ectodérmicas, o que fazemos neste trabalho. Assim sendo, estas afecções são classificadas no subgrupo 1-5, ou subgrupo tríquico e são subdivididas como segue: 1 (cabelo) divididas em: a- hipotricose, b- alopécia, c- síndrome dos cabelos anágenos frouxos (distúrbio capilar descrito nesta trabalho) e d- outros distúrbios capilares e 5 (outras afecções de origem ectodérmica), que incluem o olho (retina), divididas em: a - distrofia macular e b -degeneração retiniana. Fica o grupo B na associação entre displasia (1) e distrofia e degeneração (5), Temos no grupo B das displasias ectodérmias os seguintes subgrupos possíveis teoricamente (Tabela 2).
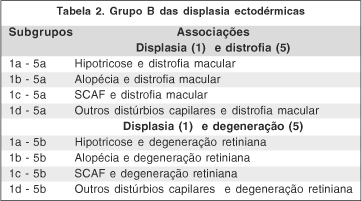
Foram incluídas nesta classificação o envolvimento somente do cabelo e retina; foram excluídas as associações com qualquer outra estrutura da ectoderme, como exemplo a síndrome EEM - displasia ectodérmica, ectrodatilia e distrofia macular(20).
A análise desta classificação mostra que a maioria dos relatos são do subgrupo 1a — 5a (hipotricose e distrofia macular), de herança autossômica recessiva (em geral de pais consangüíneos), cujos pacientes apresentaram cílios e sobrancelhas normais e hipotricose de origem congênita. Nos subgrupos 1a — 5b (hipotricose e degeneração retiniana) e 1c — 5b (SCAF e degeneração retiniana), não se encontram relatos na literatura médica até o presente momento. Fica em aberto uma possível complementação desses subgrupos no futuro, bem como outros tipos de distúrbios capilares associado à distrofia macular e degeneração retiniana (subgrupos 1d — 5a e 1d — 5b). Assim sendo, temos no grupo B das displasias ectodérmicas os seguintes subgrupos descritos (tabela 3).
CONCLUSÕES
Demonstrou-se a importância do exame dos cabelos em pacientes com distrofia macular e hipotricose congênita. A pesquisa dessas associações contribui para o diagnóstico desta entidade e foi determinado a relação direta entre distrofia macular (DRORE) e o distúrbio capilar (SCAF), ou seja, quanto pior o distúrbio capilar, mais grave é o problema ocular. Os pacientes heterozigotos, não se queixavam do crescimento anormal dos cabelos, mas apresentavam sinais da SCAF, sem apresentar a DRORE. Os pacientes homozigotos, com queixas sobre o crescimento dos cabelos, apresentavam SCAF associada à DRORE. Sugerimos que a associação da DRORE e SCAF é uma nova entidade nosológica pertencendo ao grupo B das displasias ectodérmicas. Uma nova classificação foi sugerida, englobando todos os pacientes com distúrbio capilares e distrofia e degenerações retinianas como grupo B das displasias ectodérmicas.
REFERÊNCIAS
1. Sato MT, Marzagão R, Moreira Júnior CA. Distrofia retiniana com onda rápida escotópica (DRORE) associada à síndrome dos cabelos anágenos frouxos (SCAF). Parte I: Exame oftalmológico. Arq Bras Oftalmol. No prelo 2004.
2. Brunette JR. Clinical electroretinography. Part 1: Foundations. Can J Ophthalmol. 1982;17(4):143-9.
3. Brunette JR. Clinical electroretinography. Part 2: Applications. Can J Ophthalmol. 1982;17(6):239-44.
4. Baden HP, Kvedar JC, Magro CM. Loose anagen hair as a cause of hereditary hair loss in children. Arch Dermatol. 1992;128(10):1349-53.
5. Sprecher E, Bergman R, Richard G, Lurie R, Shalev R, Petronius D, et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, enconding P-chaderin. Nat Genet. 2001;29(2):134-6.
6. Tosti A, Colombati S, De Padova MP, Guidi SG, Tosti G, Maccolini E. Retinal pigment epithelium function in alopecia areata. J Invest Dermatol. 1986;86(10):553-5.
7. Noell WK. The origin of the electroretinogram. Am J Ophthalmol. 1954;38:78-90.
8. Stühmer W, Conti F, Suzuki H, Wang X, Noda M, Yahagi N, Kubo H, Shosaku N. Structural parts involved in activation and inactivation of the sodium channel. Nature. 1989;339(6226):597-603.
9. Ganong WF. As bases gerais e celulares da fisiologia médica. In: Ganong WF, editor. Fisiologia médica. Rio de Janeiro: Prentice-Hall do Brasil; 1998, p.28-33.
10. Tripathi RC, Chalam KV, Cibis GW, Kardon RH, Tripathi BJ, Van Kujik FJGM, Weleber RG, Wand M. Retina. In: American Academy of Ophthalmology. Fundamental and principles of ophthalmology. San Francisco: American Academy of Ophthalmology; 1999. p.344-65.
11. Pagnan NAB, Gollop TR, Lederman H. The Tel-Hashomer camptodactyly syndrome: report of a new case and review of the literature; Am J Med Genet. 1988;29(2):411-7.
12. Sertie AL, Sousa AV, Steman S, Pavanello RC, Passos-Bueno MR. Linkage analysis in a large brazilian family with Van der Woude syndrome suggests the existence of a susceptibility locus for cleft palate at 11p11.2-11.1. Am J Hum Genet. 1999;65(2):433-40.
13. Murphy MF, McGinnity FG, Allen GE. New familial association between coloboma and loose anagen syndrome. Clin Genet. 1955;47:214-6.
14. Freire-Maia N. Ectodermal dysplasias. Hum Hered. 1971;21(4):309-12.
15. Freire-Maia N, Pinheiro M. Definitions and classifications. In: Freire-Maia N, Pinheiro M, editores. Ectodermal dysplasias — a clinical and genetic study. New York: Alan R Liss; 1984. p.5-25.
16. Freire-Maia N, Pinheiro M. Definitions and classifications. In: Freire-Maia N, Pinheiro M, editores. Ectodermal dysplasias — a clinical and genetic study. New York: Alan R Liss; 1984. p.25-31.
17. Kroll P. Congenital dystrophy of the pigment layer of the posterior pole with congenital total hypotrichosis. Klin Monatsbl Augenheilkd. 1981;178(2):118-20.
18. Becker M, Rohrchneider K, Tilgen W, Weber BH, Volcher HE. Familial juvenile macular dystrophy with congenital hypotrichosis capitis. Ophthalmologe. 1998;95(4):233-40.
19. Warburg M. macular dystrophy and hypotrichosis: the EEM-Albrectsen syndrome. Ophthalmic Genet.1995;16(4):177-8. Commented on: Ophthalmic Genet. 1995;16(1):11-5.
20. Ohdo S, Hirayama K, Terawaki T. Association of ectodermal dysplasia, ectrodactyly, and macular dystrophy. The EEM syndrome. J Med Genet. 1983;20(1):52-7.
21. Kroll P. Congenital dystrophy of the pigment layer of the posterior pole with congenital total hypotrichosis. Klin Mbl Augenheilk. 1981;178(2):118-20.
22. Yasukura K, Mizuta S, Maeda K. Macular degeneration in sinblings with unusual hairgrowth. Jap J Clin Ophthalmol. 1967;21:495-500.
23. Mais FAQ, Cunha MC, Cunha RP. Distrofia do epitélio pigmentar retiniano do polo posterior associada com hipotricose congênita difusa. Arq Bras Oftalmol. 1984;47(4):137-41.
24. Marren P, Wilson C, Dawber RP, Walshe MM. Hereditary hypotrichosis (Marie-Unna type) and juvenile macular degeneration (Stargardt's maculopathy). Clin Exp Dermatol. 1992;17(3):189-91.
25. Souied E, Amalric P, Chauvet ML, Chevallier C, Le Hoang P, Munnich A, et al. Unusual association of juvenile macular dystrophy with congenital hypotrichosis occurrence in two siblings suggesting autosomal recessive inheritance. Ophthalmic Genet. 1995;16(1):11-5. Commented on: Ophthalmic Genet. 1995;16(4):177-8.
26. Raison-Peyron N, Duval PA, Barneon G, Durand I, Arnaud B, Meynadier J, et al. A syndrome combining severe hypotrichosis and macular dystrophy: absence of mutations in TIMP genes. Br J Dermatol. 2000;143(4):902-4.
27. Björk A, Jahnberg P. Retinal dystrophy combined with alopecia. Acta Ophthalmol. (Copenh) 1975;53(5):781-9.
28. Johnston SS, Darragh J, Nevin NC. Hereditary macular degeneration in three generations. Br J Ophthalmol. 1973;57(8):578-83.
29. Küster W, Majewski F, Hammerstein W. Alopecia, macular degeneration, and growth retardation: a new syndrome? Am J Med Genet. 1987;28(2):477-81.
30. Da Cruz L, McAllister IL. An australian family with macular dystrophy linked to autosomal recessive alopecia universalis. Br J Ophthalmol. 2001;85(2):239.
31. Sato MT, Marzagão R, Graff C, Arana J, Moreira ATR, Pagnan NAB, Ribeiro EMSF, Moreira Júnior CA. Descrição de nova distrofia macular associada à síndrome dos cabelos anágenos frouxos. Arq Bras Oftalmol. 2002;65(2):249-56.
Endereço para correspondência
Dr. Mário Teruo Sato
Av. Winston Churchill, 1323
CEP 81130-000 Curitiba (PR)
E-mail: [email protected]
Recebido para publicação em 20.11.2003
Versão revisada recebida em 23.03.2004
Aprovação em 30.03.2004
Universidade Federal do Paraná, Centro da Visão do Hospital de Clínicas - R. Pasteur, 26 - CEP 80250-080 Curitiba (PR)
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. André Barbosa Castelo Branco

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}
{kind=link}