Arq. Bras. Oftalmol. 2004; 67 (3): 10.1590/S0004-27492004000300031
Total: 2285
Gustavo Victor1; Paloma Juni Godinho Campos2; Milton Ruiz Alves3; Walton Nosé1
DOI: 10.1590/S0004-27492004000300031
RESUMO
A cistinose é doença autossômica recessiva rara caracterizada pelo acúmulo do aminoácido cistina livre dentro dos lisossomos e geralmente é fatal na primeira década de vida na ausência de transplante renal. O presente estudo tem por objetivo relatar os achados da microscopia confocal in vivo em paciente adulto com cistinose infantil. O exame de microscopia confocal in vivo revelou que há diferenças quanto à intensidade de acometimento, tamanho e forma dos depósitos nas diversas camadas corneanas.
Descritores: Microscopia confocal; Estroma corneal; Doenças da córnea; Cistinose; Relato de caso
ABSTRACT
Cystinosis is a rare recessive autosomic disease characterized by the accumulation of the cystine amino acid inside the lysosomes, and it is generally fatal in the first decade of life in the absence of renal transplant. This study has the purpose to describe the in vivo confocal microscopy findings in a patient with infantile cystinosis. The in vivo confocal microscopy examination showed differences in intensity, size and shapes of the corneal deposits in the several corneal layers.
Keywords: Microscopy, confocal; Corneal stroma; Corneal diseases; Cystinosis; Case report
INTRODUÇÃO
A cistinose é uma doença autossômica recessiva rara caracterizada pelo acúmulo do aminoácido cistina livre dentro dos lisossomos, resultando no aparecimento de cristais de cistina na medula óssea, leucócitos, linfonodos, rins, córnea, conjuntiva, íris, corpo ciliar, esclera, coróide, epitélio pigmentar da retina, músculos extra-oculares e nervo óptico. O defeito primário está no transporte ativo de cistina através da membrana lisossomal(1-9). Uma mutação genética tem sido mapeada no cromossomo 17p(5-6). Três formas fenotípicas são descritas(1,3-4,6,10): A) Forma Infantil: é a mais comum e a mais devastadora. Também chamada de nefropática, leva ao desenvolvimento da síndrome de Fanconi. Esta forma pode conter 100 vezes mais cistina que o normal, cursa com retardo do crescimento e desenvolvimento e geralmente é fatal na primeira década de vida na ausência de transplante renal. Os sinais e sintomas oculares mais comuns são fotofobia, prurido, depósito de cistina na conjuntiva, córnea e retinopatia periférica. B) Forma Intermediária ou Adolescente: É caracterizada por nefropatia menos severa que a forma infantil, mas que compromete a expectativa da vida. As alterações evidenciadas na segunda ou terceira década de vida, apresentam-se ou não com falência renal e depósitos característicos na conjuntiva e córnea. A retinopatia não é observada. C) Forma Adulta: Esta forma benigna, que pode conter 30 vezes mais cistina que o normal, é assintomática, à exceção da fotofobia, que freqüentemente leva ao diagnóstico em exame oftalmológico de rotina. Esta forma se apresenta da adolescência à quinta década de vida. Estes pacientes têm expectativa de vida normal. Os heterozigotos têm nível de cistina 4 a 5 vezes maior que o normal, não apresentam anormalidades clínicas e têm expectativa de vida normal.
A partir de 1990, a microscopia confocal permitiu estudar a histologia de toda córnea in vivo(11-12), ajudando a compreender, diagnosticar e tratar as alterações corneanas. O objetivo deste estudo é relatar os achados da microscopia confocal in vivo em uma paciente com cistinose infantil.
RELATO DO CASO
Paciente de 19 anos, feminina, natural de Santa Catarina, Brasil. Nascida de casamento consangüíneo (pais primos), cuja cistinose infantil foi diagnosticado na infância, sendo submetida a transplante renal (1 rim) aos 6 anos de idade. Apresentava-se, à inspeção, com baixa estatura para a idade e queixava-se de intensa fotofobia e dor ocular discreta e intermitente.
O exame oftalmológico revelou: acuidade visual sem correção (AVSC) 20/20 em ambos olhos (AO). Exame refratométrico mostrou emetropia em AO e motilidade ocular extrínseca normal. A biomicroscopia revelou depósito de cristais distribuído difusamente e por toda espessura do estroma corneano. Não foram observadas alterações no epitélio, endotélio, pálpebras, conjuntiva, íris e cristalino em AO. O exame fundoscópico foi normal em AO. A pressão intra-ocular foi 14 mmHg AO às 13:20 h. Os reflexos fotomotores apresentaram-se normais em AO.
A microscopia confocal in vivo de ambos os olhos foi realizado por um dos autores (GV). O aparelho utilizado foi ConfoScan 2.0® (Fortune Technologies® Italy).
Microscopia confocal in vivo
O ConfoScan 2.0® utilizado é equipado com a lente objetiva: Achroplan 40 / 0,75 W ¥/0, que tem uma distância de trabalho de 1,98 mm(13). O seu manuseio e funcionamento já foram descritos anteriormente(11,13-16).
Ao iniciar o exame explicou-se previamente o procedimento à paciente, então foi instilada uma gota de Cloridrato de Proximetacaína 0,5% (Anestalcon®, Alcon, São Paulo, Brasil). Para a realização do exame, foi utilizada 1 gota do ácido poliacrílico a 0,002%, (Refresh Gel®, Chauvin, Audenas, França e importado pela Allergan, São Paulo, Brasil), sobre a lente objetiva. Após o correto posicionamento do paciente e do aparelho, realizou-se o exame.
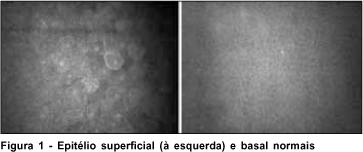
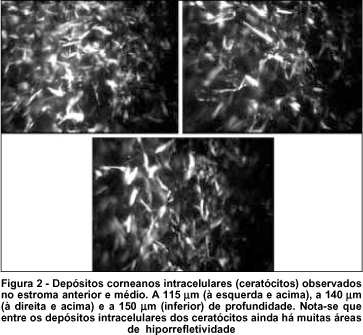
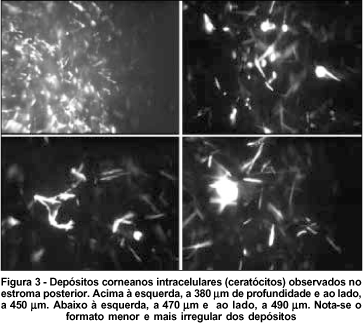
O exame da região central das córneas revelou: ausência de depósitos no epitélio superficial e basal (Figura 1), depósitos no estroma anterior e médio que se caracterizaram de alta refletividade, de formas variadas, sendo mais comum formas retangulares e fusiformes, intracelulares com a manutenção da transparência (hiporrefletividade) do compartimento extracelular (Figura 2), permitindo uma boa refração da luz através da córnea. Em menor densidade, o estroma posterior foi acometido, sendo observado formas mais irregulares, fusiformes e retangulares menores que no estroma anterior (Figura 3). Não foi observado acometimento da maior parte do endotélio, mas algumas células denunciaram o acometimento endotelial (Figura 4). O endotélio apresentou-se com leve pleomorfismo e polimegatismo e densidade de 1.940 cel/mm2 no olho direito e 1.935 cel/mm2 no olho esquerdo. Não se observou nenhum nervo corneano durante o exame em ambos olhos e a paquimetria com o microscópio confocal foi 521 e 541 mm nos olhos direito e esquerdo respectivamente.

DISCUSSÃO
A microscopia confocal permite estudar histologicamente in vivo toda córnea, sendo portanto um exame ao mesmo tempo prático, eficiente e pouco invasivo. Seu emprego a partir de 1990(11) contribuiu decisivamente para o diagnóstico, tratamento e compreensão de diversas afecções corneanas(17-21). No Brasil, os autores(14-16) realizaram os primeiros estudos com a microscopia confocal in vivo. No presente estudo, avalia-se a cistinose com seus depósitos corneanos característicos.
Já foi demostrado(22), com a microscopia confocal in vivo, que a densidade de ceratócitos em córneas normais é maior no estroma anterior diminuindo em direção posterior, onde chega a ser 30% menor. A paciente deste estudo, com 19 anos, portadora da forma infantil de cistinose, apresentou, à biomicroscopia, depósitos difusos de cristais de cisteína (tanto na periferia como no centro da córnea), e em toda profundidade do estroma, poupando epitélio e endotélio. Com a microscopia confocal foi observada densidade maior de depósitos no estroma anterior que no posterior, não acometimento do epitélio e o discreto envolvimento do endotélio corneano. Em relação à forma dos depósitos, foram observadas mais as formas retangulares e fusiformes no estroma anterior e formas mais irregulares e fusiformes menores no posterior.
O estudo(3) de um botão corneano de uma paciente de 20 anos portadora de cistinose infantil com microscopia eletrônica também revelou que o epitélio e endotélio estavam pouco afetados e todo estroma acometido, principalmente o anterior. As diferentes taxas de metabolismo dos ceratócitos nas diferentes regiões da córnea foi considerada uma possível causa para a diferença na distribuição dos depósitos no estroma. Este mesmo estudo(3) também observou que as formas dos depósitos variavam com a profundidade do estroma estudado. No estroma anterior, observou formas retangulares grandes, e no posterior, formas irregulares, retangulares pequenas, ovais e mais esparsas, achados similares ao deste estudo. Outro autor(7) observou que os cristais corneanos retangulares na cistinose são formados de L-cistina e presume-se(3) que os fusiformes são compostos de cistina, mas este ainda não tem sido provado.
Na cistinose, os característicos cristais corneanos, que se parecem com pontas de alfinete e fusiformes, não estão presentes ao nascimento(23), mas ocorrem ao redor de 1 ano de vida precedendo o início da insuficiência renal. Os cristais se depositam inicialmente na periferia e no estroma anterior corneano. Com o tempo, vão se direcionando centrípeta e posteriormente. Por volta dos 7 anos, os cristais podem ser achados em todo estroma e na maioria das vezes no endotélio. Os depósitos são simétricos, mais densos e maiores no estroma anterior(3).
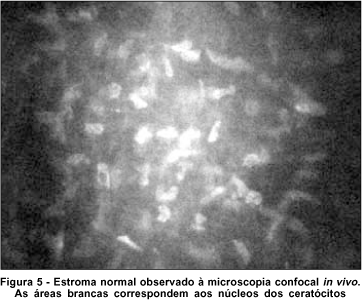
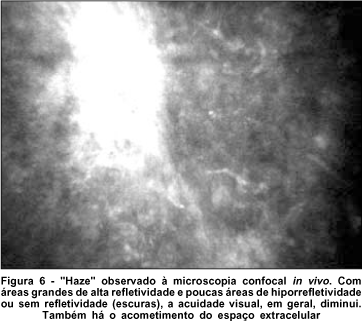
Os pacientes com cistinose desfrutam de boa acuidade visual pois as regiões entre os ceratócitos permitem a passagem da luz através da córnea como no estroma normal (Figura 5). O mesmo não acontece em outras alterações que também acometem estruturas corneanas extracelulares, como no "haze" (Figura 6), onde há bloqueio à passagem da luz através da córnea, diminuindo a acuidade visual. A expectativa de vida dos pacientes portadores de cistinose infantil tem aumentado com a realização de transplante e cuidados renais. A diminuição da função visual nestes pacientes geralmente ocorre como resultado de anormalidades da função retiniana, sinéquias posteriores, glaucoma(24) e retinopatia hemorrágica(2,25-26).
Os pacientes com cistinose infantil têm a sensibilidade corneana significativamente diminuída, com inervação pupilar e condução dos nervos trigêmios normais(4). Este autor(4) sugere que a sensibilidade corneana reduz-se por alterações funcionais no plexo neural epitelial basal e nos nervos penetrantes do estroma anterior ou em suas terminações, localizações estas, com maior concentração de depósitos de cristais corneanos(3). Dor, nestes pacientes, tem sido correlacionada aos depósitos corneanos nestas camadas(6). No caso em estudo não foi medida a sensibilidade táctil corneana. Também não foram observados nervos corneanos durante o exame confocal das córneas, fato não habitual.
A paciente apresentava espessura corneana normal em AO e contagem endotelial abaixo do normal para idade. Entretanto, há autores(5) que referem que um aumento na espessura corneana nos pacientes com esta forma de cistinose já no começo da doença, pode ser interpretada como uma disfunção subclínica do endotélio corneano. No caso em estudo pode-se observar que o endotélio de ambas as córneas apresentou algumas áreas de depósitos, polimegatismo e pleomorfismo, sugerindo acometimento endotelial.
Foi prescrito colírio de cisteamine (tiol livre) a 0,55% a cada 1h e 30 minutos (± 10 vezes ao dia) e futuramente deverá ser avaliada sua eficácia nesta paciente. Há controvérsia quanto à eficácia desta medicação. Para alguns autores(6,27-28) seu uso reduz a densidade dos depósitos cristalinos, dor e erosões corneanas. Para outros autores, não ocorrem diferenças significativas quando usadas em baixas concentrações(6). Parece que o colírio de cisteamine reage com a cistina no interior dos lisossomos formando a cisteína-cisteamine dissulfido, que parece estruturalmente com a lisina, e é transportada através do lisossomo pelo sistema de transporte da lisina, que está normal(1,6).
Com o aumento da expectativa de vida dos pacientes com cistinose infantil após transplante renal, tem-se a oportunidade de observar in vivo as alterações corneanas mais tardias desta doença.
CONCLUSÃO
A microscopia confocal empregada na avaliação deste caso mostrou-se uma importante ferramenta para o estudo in vivo dos depósitos corneanos de cisteína.
A cistinose infantil acomete mais o estroma anterior que o posterior e há diferenças na forma e tamanho dos depósitos nestas regiões da córnea. O endotélio é acometido em menor intensidade que o estroma, e o epitélio não foi acometido.
REFERÊNCIAS
1. Rosenberg LE. Doenças de armazenamento dos aminoácidos. In: Wilson JD, Braunwald E, Isselbacher KJ, Petersdorf RG, Martin JB, Fauci AS, Root RK, editors. Harrison Medicina Interna, 12ª ed. Rio de Janeiro: Guanabara Koogan; 1992. p.227-8.
2. Kaiser-Kupfer MI, Caruso RC, Minkler DS, Gahl WA. Long-term ocular manifestations of nephropathic cystinosis. Arch Ophthalmol 1986;104:706-11.
3. Melles RB, Schneider JA, Rao NA, Katz B. Spatial and temporal sequence of corneal crystal deposition in nephropathic cystinosis. Am J Ophthalmol 1987;104:598-604.
4. Katz B, Melles RB, Schneider JA. Corneal sensitivity in nephropathic cystinosis. Am J Ophthalmol 1987;104:413-6.
5. Katz B, Melles RB, Schneider JA, Rao N. Corneal thickness in nephropathic cystinosis. Br J Ophthalmol 1989;73:665-8.
6. Bridges WZ, Palay DA. Corneal deposits. In: Krachmer JH, Mannis MJ, Holland EJ, editors. Cornea. St Louis: Mosby; 1997. p.417-28.
7. Frazier PD, Wong VG. Cystinosis. Histologic and crystallographic examination of crystals in eye tissues. Arch Ophthalmol 1968;80:87-91.
8. Sanderson PO, Kuwabara T, Stark W, Wong VG, Collins EM. Cystinosis. A clinical, histopathologic and ultrastructural study. Arch Ophthalmol 1974;91:270-4.
9. François J, Hanssens M, Coppieters R, Evens L. Cystinosis. A clinical and histopathologic study. Am J Ophthalmol 1972;73:643-50.
10. Kaiser-Kupfer MI, Datiles MB, Gahl WA. Corneal transplant in a twelve-year-old boy with nephropathic cystinosis. Lancet 1987;1:331.
11. Cavanagh HD, Jester JV, Essepian J, Shields W, Lemp MA. Confocal microscopy of the living eye. CLAO J 1990;16:65-73.
12. Grupcheva CN, Craig JP, Sherwin T, McGhee CN. Differential diagnosis of corneal oedema assisted by in vivo confocal microscopy. Clin Experiment Ophthalmol 2001;29:133-7.
13. Confoscan 2.0. Full computerized confocal microscope, specifications and operating manual. Greensboro NC: Fortune Technologies.
14. Victor G, Nosé W. Microscopia confocal in vivo em ceratocone: relato de caso. Rev Bras Oftalmol 2001;60:853-8.
15. Victor G, Alberti GN, Nosé W. Microscopia confocal in vivo na epitelização da interface após LASIK. Rev Bras Oftalmol 2002;61:284-9.
16. Victor G, Alves MR, Nosé W. Avaliação in vivo da histologia corneana após ceratotomia radial com a microscopia confocal. Rev Bras Oftalmol 2003;62:641-51.
17. Li HF, Petroll WM, Moller-Pedersen T, Maurer JK, Cavanagh HD, Jester JV. Epithelial and corneal thickness measurements by in vivo confocal microscopy through focusing (CMTF). Curr Eyes Res 1997;16:214-21.
18. Prydal JL, Franc F, Dilly PN, Kerr Muir MG, Corbett MC, Marshall J. Keratocyte density and size in conscious humans by digital image analysis of confocal images. Eye 1998;12(Pt 3A):337-42.
19. Tomii S, Kinoshita S. Observations of human corneal epithelium by tandem scanning confocal microscope. Scanning 1994;16:305-6.
20. Cavanagh HD, Petroll WM, Alizadeh H, He YG, McCulley JP, Jester JV. Clinical and diagnostic use of in vivo confocal microscopy in patients with corneal diseases. Ophthalmology 1993;100:1444-54.
21. Cogan DG, Kuwabara T. Ocular pathology of cystinosis. Arch Ophthalmol 1960;63:51-7.
22. Trauner DA, Chase C, Scheler J, Katz B, Schneider JA. Neurologic and cognitive deficits in children with cystinosis. J Pediatr 1988;112:912-4.
23. Mustonen RK, McDonald MB, Srivannaboon S, Tan AL, Doubrava MW, Kim CK. Normal human corneal cell populations evaluated by in vivo scanning slit confocal microscopy. Cornea 1998;17:485-92.
24. Goldsmith LA. Cutaneous changes in errors of amino acid metabolism: alkaptonuria. In: Fitzpatrick TB, Eisen AZ, Wolff K, editors. Dermatology in general medicine. 3rd ed. New York: McGraw-Hill; 1987. p.1642-6.
25. Katz B, Melles RB, Schneider JA. Recurrent crystal deposition after keratoplasty in nephropathic cystinosis. Am J Ophthalmol 1987;104:190-1.
26. Gahl WA, Reed GF, Thoene JG, Schulman JD, Rizzo WB, Jonas AJ, et al. Cysteamine therapy for children with nephropathic cystinosis. N Engl J Med 1987;316:971-7.
27. Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab 2000;71:100-20.
28. Iwata F, Kuehl EM, Reed GF, McCain LM, Gahl WA, Kaiser-Kupfer MI. A randomized clinical trial of topical cysteamine disulfide (cystamine) versus free thiol (cysteamine) in the treatment of corneal cystine crystals in cystinosis. Mol Genet Metab 1998;64:237-42.
Endereço para correspondência
Dr. Gustavo Victor
Av. República do Líbano, 1034
São Paulo (SP)
CEP 04502-001
E-mail: [email protected]
Recebido para publicação em 11.07.2003
Versão recebida em 27.02.2004
Aprovação em 18.03.2004

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.