Arq. Bras. Oftalmol. 2003; 66 (5): 10.1590/S0004-27492003000500024
Total: 2667
Jacó Lavinsky1; Raquel Goldhardt; Samara Keli Ariente; Cláudia G. Domingues; Fábio Lavinsky
DOI: 10.1590/S0004-27492003000500024
RESUMO
A síndrome de Bardet-Biedl é doença de herança autossômica recessiva caracterizada por distrofia retiniana, polidactilia, obesidade, retardo mental e hipogenitalismo. Um ou mais dos achados acima que caracterizam a síndrome podem estar ausentes, mas a distrofia retiniana é achado consistente. Esta se manifesta clinicamente na infância com progressiva perda visual, causando grave dificuldade visual na adolescência. Os autores descrevem dois casos de síndrome de Bardet-Biedl, os resultados da acuidade visual, biomicroscopia, oftalmoscopia, angiografia, campo visual e eletrorretinograma. Foi realizada revisão bibliográfica com ênfase na identificação dos sinais sistêmicos, envolvimento ocular, testes eletrofisiológicos e avaliação genética.
Descritores: Síndrome de Bardet-Biedl; Retinite pigmentosa; Coróide; Criança; Adulto; Relato de caso; Literatura de revisão
ABSTRACT
Bardet-Biedl syndrome is a hereditary autosomal recessive disease characterized by retinal dystrophy, polydactyly, obesity, mental retardation, and hypogenitalism. One or more of the clinical features characterizing the syndrome may be absent, but retinal dystrophy is a consistent finding. It becomes clinically manifest in early childhood, with progressive loss of visual function, leading to severe visual disability in early adolescence. The authors describe two cases of Bardet-Biedl syndrome, the results of visual acuity, slit-lamp examination, ophthalmoscopy, angiography, visual fields and electroretinograms. The literature was reviewed stressing the importance of identifying systemic signs, ocular involvement, electrophysiologic tests and genetic evaluation.
Keywords: Bardet-Biedl syndrome; Retinitis pigmentosa; Choroid; Child; Adult; Case report; Literature review
INTRODUÇÃO
Em 1866, Laurence e Moon descreveram uma condição genética na qual os pacientes exibiam retardo mental, baixa estatura, hipogenitalismo, ataxia, paraplegia espástica e nistagmo(1). Alguns apresentavam retinose pigmentar, enquanto outros, tinham atrofia coroidiana. Mais tarde, Bardet e Biedl descreveram, independentemente, uma síndrome que consistia em obesidade, polidactilia, retinose pigmentar, retardo mental e atresia anal(2-3). Pouco tempo depois, Solis-Cohen e Weiss relataram a Síndrome de Laurence-Moon-Biedl acreditando que esta seria uma desordem única com achados semelhantes aos descritos pelos outros autores(4).
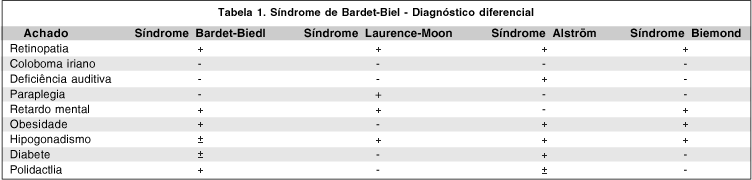
Mais recentemente sugeriu-se que as síndromes de Laurence-Moon e Bardet-Biedl são entidades clínicas diferentes (Tabela 1), com alguns achados em comum, incluindo retardo mental, hipogenitalismo, obesidade, retinopatia e herança autossômica recessiva(5-6). Polidactilia e obesidade são achados raros na síndrome de Laurence-Moon, entretanto, são encontradas com alta freqüência na síndrome de Bardet-Biedl. Na síndrome de Laurence-Moon há envolvimento neurológico progressivo, caracterizado principalmente por ataxia e paraplegia espástica. Complicações neurológicas são extremamente incomuns na síndrome de Bardet-Biedl. Os achados fundoscópicos também diferem. Na síndrome de Laurence-Moon há extensa atrofia coroidiana, semelhante à coroideremia. Todavia, na síndrome de Bardet-Biedl a retinopatia assemelha-se com a retinose pigmentar, havendo atrofia coriocapilar somente nos casos avançados(6-8). Os critérios diagnósticos da síndrome de Bardet-Biedl incluem distrofia retiniana, obesidade, polidactilia, retardo mental e hipogenitalismo. Stigglebout considera que quatro destes achados são necessários para se firmar o diagnóstico(9). Esta síndrome é absolutamente rara, com uma prevalência que varia de 1:17.500 a 1:160.000.
No presente trabalho descrevem-se dois casos com achados compatíveis com o diagnóstico de síndrome de Bardet-Biedl.
RELATO DE CASOS
Caso 1: JES, 12 anos, masculino, branco, estudante, natural de Porto Alegre/RS e procedente de Esteio. Foi encaminhado ao Serviço de Oftalmologia do Hospital de Clínicas de Porto Alegre pelo Serviço de Genética Médica do mesmo hospital. Apresentava história de baixa acuidade visual em ambos os olhos, observada primeiramente aos 2 anos de idade pelo pediatra. Nesta época lhe foram prescritos óculos; entretanto, a perda visual foi progressiva. O paciente apresentava ainda polidactilia, que foi corrigida cirurgicamente com 1 ano de idade, retardo mental e hipogonadismo. A história mórbida pregressa relata nascimento com 7 meses, pesando 1.800 g. Bolsa rota por 4 dias, infecção intraparto. Paciente ficou 1 mês no berçário. Sem outras alterações sistêmicas durante seu desenvolvimento. A história familiar mostrava pais não consangüíneos, 3 primas paternas com retardo mental e uma prima materna falecida por problema cardíaco.
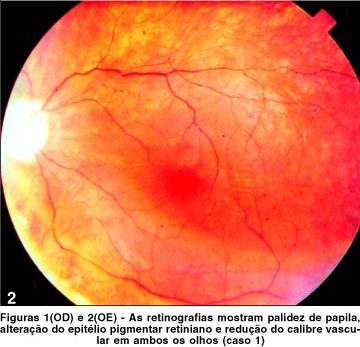
Ao exame físico, pressão arterial normal, obesidade e hipogonadismo. Ao exame oftalmológico constatou-se acuidade visual de contar dedos a 1 metro com correção para miopia (OD -6,25 esf » -1,00 cil a 45º e OE -5,50 esf » -0,75 cil a 270º) em ambos os olhos. A pressão intra-ocular, a motilidade ocular e a biomicroscopia eram normais. À fundoscopia, observou-se palidez de papila, alteração do epitélio pigmentar retiniano e redução do calibre vascular (Figuras 1 e 2) de ambos os olhos (AO).

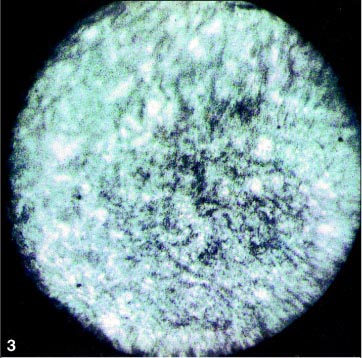
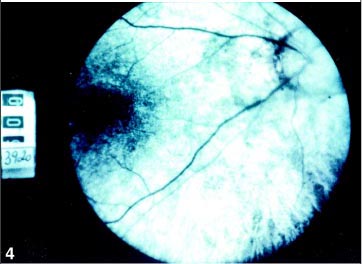
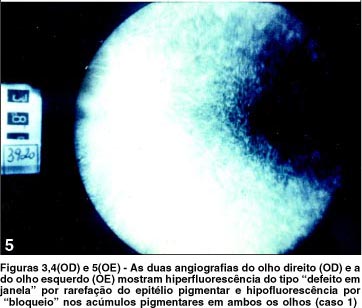
O estudo angiofluoresceínico mostrou diminuição do diâmetro vascular e alterações pigmentares sugestivas de retinose pigmentar (Figuras 3, 4 e 5) em AO. O campo visual mostrou-se tubular em AO e o eletrorretinograma evidenciou ausência de registro de atividade bilateral.
As provas de função renal, sódio e potássio séricos, glicemia, testosterona, hormônio folículos-estimulantes (FSH) e luteinizante (LH), apresentaram todos resultados normais.
Caso 2: MES, 36 anos, feminino, branca, natural e procedente de Viamão/RS. Apresentava baixa acuidade visual central e periférica percebida aos 6 anos. A paciente também apresentava polidactilia nos membros inferiores, a qual foi corrigida cirurgicamente aos dois anos de idade, bem como apêndices nas mãos, os quais foram retirados cirurgicamente dois dias após o parto. Além desses achados, foram constatados problemas digestivos, hipertensão, discreto retardo mental, obesidade e ciclo menstrual normal. Na história obstétrica, consta gestação a termo com parto natural. A história familiar registra pais não consangüíneos, irmão falecido aos 14 anos com polidactilia, cardiopatia e estrabismo. Outros dois irmãos normais. Sobrinho com imperfuração anal, sem esfíncter. Outros dois sobrinhos normais.
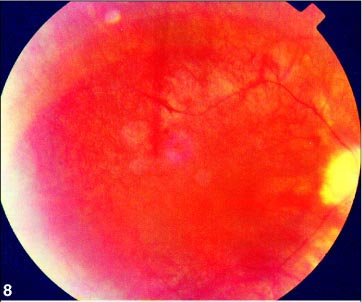
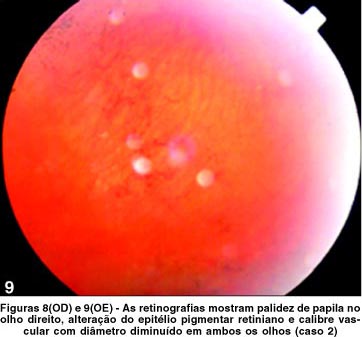
Ao exame físico, apresentou hipertensão, obesidade e cicatrizes da correção da hexadactilia de mãos e pés. Ao exame oftalmológico, constatou-se acuidade visual de 20/400 no OD e de contar dedos a 1 metro no OE com correção para astigmatismo misto (OD +0,50 » -2,00 a 5º e OE +0,50 » -1,50 a 175º) em ambos os olhos. A pressão intra-ocular era normal. A motilidade ocular apresentou rotações com hiperfunção de reto lateral esquerdo, e à biomicroscopia, catarata sub-capsular posterior bilateral e câmara anterior de média profundidade. À fundoscopia, observou-se palidez de papila, alteração do epitélio pigmentar retiniano (Figuras 8 e 9), evidenciado na angiografia pelas rarefações e acúmulos pigmentares, com hiperfluorescência nos locais de rarefação pigmentar e hipofluorescência por bloqueio nos locais com acúmulos pigmentares e redução do calibre vascular de ambos os olhos.
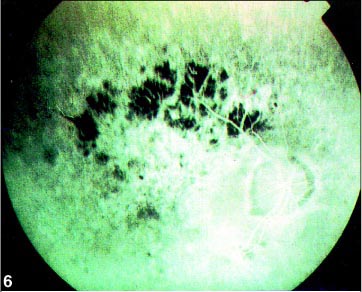
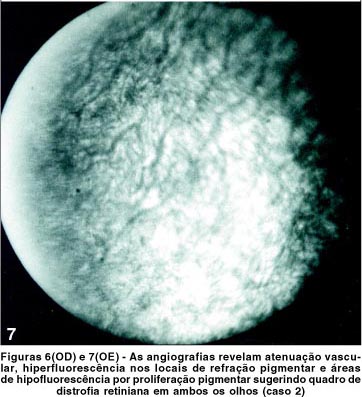
O estudo angiofluoresceinográfico mostrou ainda extensa alteração pigmentar no pólo posterior e periferia, associada à vasoconstrição, sugestiva de distrofia retiniana (Figuras 6 e 7). O campo visual mostrou-se tubular em AO, e o eletrorretinograma ausência de registro de atividade bilateral.
DISCUSSÃO
A síndrome, uma herança autossômica recessiva, é heterogênea com pelo menos quatro loci genéticos (BBS1-4) já mapeados até o momento(10), com formas de apresentação completas muito raras(5). O caso 1 tem história familiar de três primas paternas com retardo mental, e no caso 2, um irmão falecido aos 14 anos com polidactilia, cardiopatia e estrabismo.
Outras características fundoscópicas descritas incluem palidez de disco óptico, depósitos pigmentares esparsos e, mais raramente, pigmentos tipo "células ósseas" (11), além do estreitamento arteriolar. Hipopigmentação macular com atrofia do epitélio pigmentar freqüentemente ocorre num padrão em alvo. Nos dois casos descritos, observou-se palidez de papila, alteração do epitélio pigmentar retiniano e redução do calibre vascular de ambos os olhos. A angiografia fluoresceínica demonstra bem as alterações fundoscópicas descritas. O estudo fluoresceinográfico no caso 1 apresentava alterações pigmentares sugestivas de retinose pigmentar, e no caso 2, extensa alteração pigmentar no pólo posterior e periferia associada à atenuação vascular sugestiva de distrofia retiniana. O envolvimento fundoscópico mais característico da síndrome é uma distrofia retiniana pigmentar atípica com envolvimento macular precoce, podendo-se observar em casos severos membrana neovascular pré-retiniana formada a partir de estímulos angiogênicos pela má perfusão capilar, com exsudação a partir de capilares paramaculares.
Na maioria dos casos, a eletrorretinografia (ERG) é anormal, sendo o principal exame diagnóstico na retinopatia da síndrome de Bardet-Biedl. Numa série de casos acompanhados na Noruega, 36 pacientes foram submetidos a ERG que foi anormal em todos os indivíduos(12). Na presença de um ERG alterado, mesmo com exame fundoscópico normal, pode-se firmar o diagnóstico de síndrome de Bardet-Biedl em casos suspeitos(13). A falta de correlação entre a máxima resposta do ERG e as áreas residuais do campo visual, diferente do que acontece com a retinose pigmentar em pacientes não-sindrômicos, pode estar relacionada tanto com a baixa confiabilidade do teste de campo visual dos pacientes com a síndrome, como com o mecanismo de degeneração retiniana progressiva, que é diferente da retinose pigmentar pura(14). Nos dois casos estudados não houve registro de atividade bilateral.
Ao contrário da retinose pigmentar típica, a retinopatia da síndrome de Bardet-Biedl afeta a acuidade visual (AV) precocemente(6,8). A diminuição da AV aparece durante a segunda e terceira décadas de vida(9,15). Cegueira noturna, em geral, está usualmente presente. Uma baixa acuidade visual pode ser devida à má performance por déficit cognitivo. Leys et al. sugerem que os testes de adaptação ao escuro são mais sensíveis do que a medição da AV e, que o exame eletrorretinográfico é mais sensível do que o teste de adaptação ao escuro na avaliação de danos progressivos à função retiniana(16). Nos dois casos os pacientes apresentavam cegueira legal (AV < 20/400).
O campo visual começa a apresentar alterações por volta dos 10 anos de idade, e aos 24 anos, certamente, não resta mais que uma ilha de visão central. Nos dois casos a campimetria mostrou campo tubular. Já no que diz respeito à diminuição da acuidade visual, não foi estabelecida uma relação direta com a idade(17).
Também foram descritos na literatura casos de nistagmo, catarata, atrofia de íris e coloboma de íris(6). No caso 2 foi observada catarata subcapsular posterior bilateral e a motilidade ocular apresentou rotações com hiperfunção de reto lateral esquerdo.
Não há dúvidas de que a função dos bastonetes como a dos cones são afetadas na síndrome de Bardet-Biedl. Contudo, não se determinou qual o tipo de disfunção é mais precoce (distrofia dos bastonetes, distrofia dos cones ou ambas). Jacobson et al. sugerem que há maior perda da sensibilidade dos bastonetes para uma certa perda da sensibilidade dos cones(17).
Obesidade está presente em grau moderado em cerca de 90% dos pacientes. Ambos os casos estudados eram obesos.
Sindactilia e braquidactilia são achados considerados equivalentes à polidactilia. Dismorfias de extremidades ocorrem em mais de 90% dos afetados(15,18). Estes achados podem passar despercebidos em casos já corrigidos cirurgicamente e cujas cicatrizes não são aparentes. As alterações do esqueleto são as únicas detectáveis desde o nascimento(13). Os dois casos apresentaram hexadactilia de mãos e pés.
O hipogenitalismo é freqüentemente presente em homens(5,15,18). Não existem evidências laboratoriais de hipofunção hipotalâmica-hipofisária, sendo o hipogenitalismo masculino, provavelmente, um distúrbio primário da síndrome genética. O caso 1 apresentou hipogenitalismo. Até o presente momento, não há referência de um homem com síndrome de Bardet-Biedl ter gerado prole(18-19). Todavia, o hipogenitalismo é usualmente menos aparente nas mulheres do que nos homens. As mulheres, entretanto, freqüentemente apresentam evidências endócrinas de disfunção reprodutiva, sendo algumas férteis(18).
A baixa prevalência de lesões renais na síndrome de Bardet-Biedl encontrada em estudos clínicos(9,15) contrasta com a alta incidência de doença urogenital detectada nas necropsias(20). A uremia e/ou complicações hipertensivas podem ser uma das causas de morte precoce na síndrome de Bardet-Biedl. O tratamento cirúrgico precoce associado ao controle vigoroso da hipertensão pode melhorar o prognóstico(21).
Anormalidades do metabolismo da glicose como intolerância aos carboidratos e diabete mellitus dos tipo I e II, têm sido descritas entre os afetados(4,22-24). Portanto, pacientes portadores de síndrome de Bardet-Biedl devem ser rastreados para detectar a presença de alterações no metabolismo da glicose.
Existem outras síndromes, além da de Bardet-Biedl, que apresentam combinações de achados como defeitos oculares, retardo mental, hipoplasia genital, obesidade e anomalias digitais. Embora menos freqüentes, as síndromes de Laurence-Moon, Alstrom e Biemond II devem ser incluídas no diagnóstico diferencial da síndrome de Bardet-Biedl(6).
REFERÊNCIAS
1. Laurence JZ, Moon RC. Four cases of retinitis pigmentosa occurring in the same family and accompanied by general imperfection of development. Ophthalmic Rev 1866;2:32-41.
2. Bardet G. Sur un syndrome d´obésité congénitale avec polydactylie et rétinite pigmentaire (contribution à l´étude des formes cliniques de l´obésite hypophysaire). . Paris 1920. p.107.
3. Biedl A. Ein geschwisterpaar mit adioposo-genitaler dystrophie. Dtsch Med Wschr 1922;48:1630.
4. Solis-Cohen S, Weiss E. Dystrophia adipogenitalis with atypical retinitis pigmentosa and mental deficiency: the Laurence-Biedl syndrome: a report of four cases in one family. Am J Med Sci 1925;169:489-505.
5. Ammann F. Investigations cliniques et génétiques sur le syndrome de Bardet- Biedl en Suisse. J Genet Hum 1970;18(suppl):1-310.
6. Bergsma DR, Brown KS. Assessment of ophthalmologic, endocrinologic and genetics findings in the Bardet-Biedl syndrome. Birth Defects Orig Artic Ser 1975;11:132-6.
7. Schachat AP, Maumenee IH. Bardet-Biedl syndrome and related disorders. Arch Ophthalmol 1982;100:285-8.
8. Campo RV, Aaberg TM. Ocular and systemic manifestations of the Bardet-Biedl syndrome. Am J Ophthalmol 1982;94:750-6.
9. Stigglebout W. The Bardet-Biedl syndrome: including Hutchinson-Laurence-Moon syndrome. In: Vinkin PJ, Bruyn GW, editors. Neuroretinal degenerations: Handbook of clinical neurology. Amsterdam: North-Holland; 1972. p.380-412.
10. Beales PL, Warner AM, Hitman GA, Thakker R, Flinter FA. Bardet-Biedl syndrome: a molecular and phenotypic study of 18 families. J Med Genet 1997;34:92-8.
11. Fulton AB, Hansen RM, Glynn RJ. Natural course of visual functions in the Bardet-Biedl syndrome. Arch Ophthalmol 1993;111:1500-6.
12. Riise R, Andréasson S, Wright AF, Tornqvist K. Ocular findings in the Laurence-Moon-Bardet-Biedl syndrome. Acta Ophthalmol Scand 1996;74:612-7.
13. Steiner PA. Bardet-Biedl syndrome. J Am Optom Assoc 1990;61:852-5.
14. Iannaccone A, Vingolo EM, Rispoli E, De Propris G, Tanzilli P, Pannarale MR. Electroretinographic alterations in the Laurence-Moon-Bardet-Biedl phenotype. Acta Ophthalmol Scand 1996;74:8-13.
15. Bell J. The Laurence-Moon syndrome. In: Penrose LS, ed. The treasury of human inheritance. Part III. Vol. 5. London: Cambridge University Press, 1958:51-96.
16. Leys MJ, Schreiner LA, Hansen RM, Mayer DL, Fulton AB. Visual acuities and dark-adapted thresholds of children with Bardet-Biedl syndrome. Am J Ophthalmol 1988;106:561-9.
17. Jacobson SG, Borruat FX, Apáthy PP. Patterns of rod and cone dysfunction in Bardet-Biedl syndrome. Am J Ophthalmol 1990;109:676-88.
18. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G et al. The cardinal manifestation of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 1989;321:1002-9.
19. Nadjmi B, Flanagan MJ, Christian JR. Laurence-Moon-Biedl syndrome, associated with multiple genitourinary tract anomalies. Am J Dis. Child 1969;117:352-6.
20. Hurley RM, Dery P, Norady MB, Drummond KN. The renal lesion of the Laurence-Moon-Biedl syndrome. J Pediatr 1975;87:206-9.
21. Churchill DN, Mcmanamon P, Hurley RM. Renal disease - a sixth cardinal feature of the Laurence-Moon-Biedl syndrome. Clin Nephrol 1981;16:151-4.
22. Escallon F, Traboulsi EI, Infante R. A family with the Bardet-Biedl syndrome and diabetes mellitus. Arch Ophthalmol 1989;107:855-7.
23. Bruford EA, Riise R, Teague PW, Porter K, Thomson KL, Moore AT et al. Linkage mapping in 29 Bardet-Biedl syndrome families confirms loci in chromosomal regions 11q13, 15q22.3-q23, and 16q21. Genomics, 1997;41: 93-9;41:93-9.
24. Bek T, Rosenberg T. Clinical pathology and retinal vascular structure in the Bardet-Biedl syndrome. Br J Ophthalmol 1995;79:76-80.
Endereço para correspondência
Samara Keli Ariente
Rua Laurindo, 186/306
Porto Alegre (RS) CEP 90040-140
E-mail: [email protected]
Recebido para publicação em 03.06.2002
Aceito para publicação em 22.01.2003
Este trabalho foi realizado no Serviço de Oftalmologia do Hospital de Clínicas de Porto Alegre da Faculdade de Medicina da Universidade Federal do Rio Grande do Sul - UFRGS.
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos ao Dr. Sérgio Lustosa da Cunha.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}