Arq. Bras. Oftalmol. 2003; 66 (2): 10.1590/S0004-27492003000200021
Total: 3051
Leandro Cabral Zacharias1; Alexandre Marques Rosa; Yoshitaka Nakashima1; Sérgio Lustosa da Cunha1
DOI: 10.1590/S0004-27492003000200021
RESUMO
A Síndrome de Aicardi é afecção neurorretiniana de etiologia desconhecida. Seu diagnóstico baseia-se no achado de convulsões infantis e lacunas coriorretinianas associadas a alterações radiológicas características (agenesia do corpo caloso). Todos os casos relatados até o momento pertencem ao sexo feminino, com exceção de um que apresentava cariótipo 47XXY; a sobrevida até a adolescência é rara. Objetivo do presente trabalho é descrever um caso de síndrome de Aicardi, sendo provavelmente o primeiro registro desta doença em nosso meio. As lacunas coriorretinianas são elemento essencial ao diagnóstico desta síndrome, sendo consideradas patognomônicas em criança do sexo feminino apresentando convulsões. Por este motivo, cabe ao oftalmologista papel de destaque no diagnóstico desta síndrome.
Descritores: Anormalidades do olho; Corpo caloso; Síndrome; Espasmos infantis; Retina; Imagem por ressonância magnética; Tomografia computadorizada por raios X; Relato de caso
ABSTRACT
Aicardi Syndrome is a neuroretinal disorder of unknown etiology. The diagnosis is based on the finding of infantile seizures and chorioretinal lacunae associated with characteristic radiological alterations (agenesy of the corpus callosum). Till the present, all reported cases are in females, except one in a boy with a 47XXY karyotype; the children usually die before puberty. The objective is to describe a case of Aicardi syndrome. Chorioretinal lacunae are very important findings in this syndrome, considered pathognomonic in a young girl presenting seizures. The ophthalmologist has an important role in confirming the diagnosis of this syndrome.
Keywords: Eye abnormalities; Corpus callosum; Syndrome; Infantile spasms; Retina; Magnetic resonance imaging; X-ray computed tomography; Case report
INTRODUÇÃO
Em 1965, Aicardi et al(1-2) descreveram uma síndrome que consistia em: espasmos infantis, agenesia do corpo caloso e lacunas coriorretinianas. Desde então, vários casos semelhantes foram relatados, sendo que todos os indivíduos acometidos pertenciam ao sexo feminino, exceto um paciente com síndrome de Klinefelter (cariótipo 47XXY)(3). Estudos genéticos mostram possível localização do gene em Xp22(4).
As lacunas coriorretinianas são achados patognomônicos desta síndrome, de modo que o oftalmologista tem papel importante no diagnóstico desta afecção neuro-ocular.
O objetivo do presente estudo é relatar um caso de síndrome de Aicardi em que o diagnóstico foi obtido através do exame fundoscópico, sendo a nosso ver o primeiro registro desta entidade na literatura oftalmológica brasileira.
RELATO DE CASO
MSS, nove meses, feminino, nascida de parto cesáreo (pela realização de laqueadura no mesmo ato cirúrgico) com peso de 3200g.
Com dois meses de idade, começou a apresentar convulsões, sendo acompanhada na Clínica Neurológica desde então com o diagnóstico de síndrome de West e com duas a três crises diárias de espasmos em flexão, apesar do uso de anticonvulsivantes (vigabatrina 1 comprimido ao dia e fenobarbital 30 gotas ao dia). A mãe percebeu que a paciente não seguia objetos com quatro meses de idade, procurando então avaliação oftalmológica.
Ao exame oftalmológico, a paciente seguia objetos luminosos, reagindo com irritabilidade à oclusão do olho direito.
Apresentava pupilas isocóricas, reagentes, com reflexo fotomotor direto diminuído em ambos os olhos, e sem defeito aferente relativo.
A refração sob cicloplegia foi de: OD +3,00 DE –0,50 DCX180 e OE +2,00 DE.
Não foram evidenciadas alterações do segmento anterior.
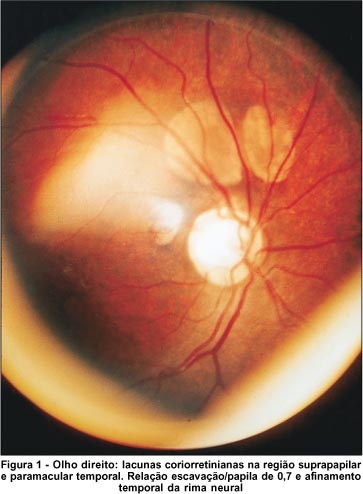
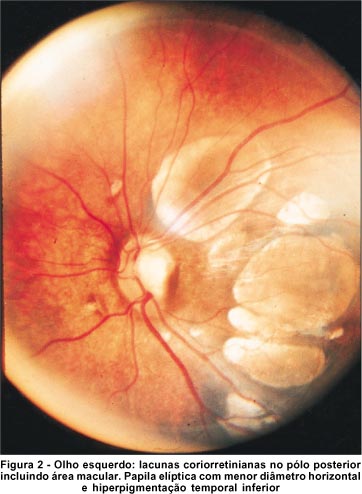
A fundoscopia mostrou lacunas coriorretinianas em ambos os olhos. No olho direito, limitavam-se à região suprapapilar e temporal à papila e variavam de 0,5 a 1,2 diâmetros papilares (Figura 1). No olho esquerdo, apresentavam-se dispostas temporalmente à papila, acometendo inclusive a área macular, variando de 0,1 a 1,2 diâmetros papilares (Figura 2). A papila do olho direito era arredondada, com escavação de 0,6x0,7. No olho esquerdo, era elíptica, com diâmetro horizontal reduzido, apresentando hiperpigmentação temporal inferior e escavação de 0,4x0,6.
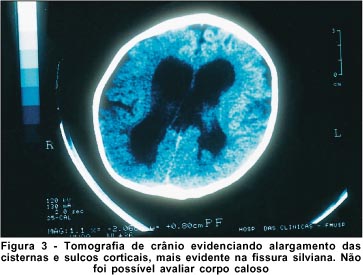
A tomografia computadorizada do crânio evidenciou alargamento das cisternas e sulcos corticais, mais evidente na fissura silviana. Não foi possível avaliar o corpo caloso por limitações do método (Figura 3).
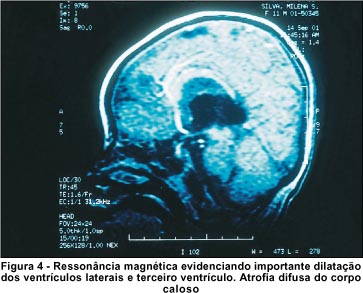
A ressonância nuclear magnética do crânio mostrou dilatação importante dos ventrículos laterais e terceiro ventrículo, não havendo desvio das estruturas em relação à linha média. A análise do corpo caloso evidenciou que o mesmo estava presente, mas com aparência hipoplásica (Figura 4).
O exame de eletroencefalograma revelou onda lenta difusa de atividade rápida assimétrica em quadrantes posteriores; atividade epileptiforme multifocal, com projeções posterior, frontal e temporal(5-6).
DISCUSSÃO
A síndrome de West tem início nos primeiros meses de vida e apresenta como manifestações clínicas repentinas e rápidas aduções e flexões de membros com concomitante flexão de cabeça e tronco. Os espasmos podem ocorrer em salvas, com a criança dormindo, andando ou sendo manipulada.
Uma ampla gama de patologias compõem o diagnóstico diferencial da síndrome de West: malformações pré-natais, lesão central por hipóxia ou hipoglicemia, alterações bioquímicas (como deficiência de piridoxina e acidúria), causas infecciosas (como toxoplasmose ou citomegalovírus), esclerose tuberosa e síndrome de Aicardi.
A síndrome de Aicardi é um distúrbio cerebroretiniano de etiologia desconhecida. O diagnóstico desta síndrome é estabelecido quando espasmos infantis e lacunas coriorretinianas ocorrem em combinação com alterações neurológicas e eletrofisiológicas estabelecidas. O achado oftalmoscópico característico da síndrome de Aicardi consiste em áreas redondas, hipopigmentadas, não elevadas, denominadas lacunas coriorretinianas. Estas lesões são brancas ou amareladas e apresentam limites nítidos, em geral pouco pigmentadas, agrupando-se ao redor do nervo óptico. Variam de tamanho entre 0,1 a mais de cinco diâmetros papilares e normalmente são bilaterais(1-2), havendo porém casos unilaterais relatados(7). A ausência de pigmento dentro das lacunas coriorretinianas auxilia no diagnóstico diferencial com coriorretinites infecciosas(8). As lacunas coriorretinianas já foram descritas em associação com outras entidades raras, incluindo: a síndrome da banda amniótica(9), a síndrome oro-digital-facial tipo VIII (10) e em uma forma dominante rara de microcefalia(11). Entretanto, em criança do sexo feminino com convulsões, o achado de múltiplas lacunas coriorretinianas peripapilares é considerado patognomônico da síndrome de Aicardi(1-2,12-13).
Histologicamente, as lacunas coriorretinianas consistem em defeitos limitados ao epitélio pigmentado da retina e hipopigmentação da coróide. A retina que recobre as lacunas apresenta alterações histológicas, como atrofia da retina externa e conseqüente degeneração secundária dos fotorreceptores(8).
O diâmetro da maior lacuna relaciona-se com o desenvolvimento neurológico: quanto menor a lacuna, melhor o prognóstico quanto ao desenvolvimento de deambulação e de fala. Esta correlação não foi observada com a acuidade visual, que se associa com a localização, e não com o tamanho da lacuna; um mau prognóstico visual relaciona-se com o envolvimento da fóvea. Outros fatores relacionados com mau prognóstico visual são: descolamento da retina, anisometropia, e alterações corticais resultantes de sedação por anticonvulsivantes, convulsões intratáveis e malformações anatômicas(12).
Além das lacunas coriorretinianas, outras malformações oculares podem ser observadas: coloboma e hipoplasia do nervo óptico, pigmentação congênita do disco óptico, microftalmia, persistência de membrana pupilar, cisto retrobulbar, pseudoglioma, descolamento da retina, cicatrizes maculares, catarata, sinéquias e colobomas da íris(12).
Exames de neuroimagem podem revelar a agenesia parcial ou completa do corpo caloso, dilatação ventricular, hidrocefalia, e heterotopia cortical(13-14).
Acredita-se que esta síndrome tenha herança dominante ligada ao X, sendo letal em indivíduos do sexo masculino nos primeiros meses de gestação. Uma vez que todos os casos descritos não possuem história familiar e o status heterozigoto associa-se com a expressão da doença, supõe-se que todos os casos sejam resultantes de mutações novas e o risco de um novo caso em futuras gestações seria raro e igual ao risco de uma nova mutação(4,15).
Apesar de ter etiologia desconhecida, o momento da embriogênese em que ocorrem as alterações nesta síndrome pode ser inferido. O epitélio pigmentado da retina forma-se na quarta, e a pigmentação da coróide na quinta semana de gestação, simultaneamente ao fechamento da papila óptica. Considerando-se os achados histológicos da síndrome de Aicardi, supõe-se que o fator patogênico envolvido deva exercer sua ação entre a quarta e a quinta semanas do crescimento fetal(7).
Espasmos infantis são normalmente a primeira manifestação da síndrome de Aicardi, apresentando-se clinicamente por contrações musculares rápidas, com hiperextensão da cabeça, flexão ou extensão do tronco e braços, episódios estes múltiplos ao longo do dia e com início no primeiro ano de vida. As associações sistêmicas mais comuns na síndrome de Aicardi são malformações vertebrais e da coluna (vértebras fundidas, escoliose, espina bífida) e de costela (costela ausente, adicional ou bifurcada)(16).
A sobrevida até a adolescência é rara, sendo que o óbito é normalmente resultante de infecções pulmonares. O prognóstico de desenvolvimento neuro-psico-motor é reservado devido ao retardo mental profundo e convulsões intratáveis(12).
CONCLUSÃO
As lacunas coriorretinianas são elementos essenciais no diagnóstico desta síndrome, sendo consideradas patognomônicas em criança do sexo feminino apresentando convulsões (espasmos musculares). Por este motivo, cabe ao oftalmologista papel de destaque no diagnóstico da síndrome de Aicardi.
REFERÊNCIAS
1. Aicardi J, Lefebvre J, Lerique-Koechlin A. A new syndrome: spasm in flexion, callosal agenesis, ocular abnormalities. Eletroencephalogr Clin Neurophysiol 1965;19:606-12.
2. Aicardi J, Chevrie JJ, Rousselie F. Le syndrome spasmes em flexion, agenesie calleuse, anomalies chorio-rétiniennes. Arch Fr Pediatr 1969;26:1103-20.
3. Hopkins IJ, Humphrey I, Keith CG, Susman M, Webbg C, Turner EK. The Aicardi syndrome in a 47, XXY male. Aust Pediatr J 1979;15:278-80.
4. OMIM. Online Medelian Inheritance in Man. Corpus callosum, agenesis of, with chorioretinal abnormality. Online Mendelian Inheritance in Man (OMIN) Johns Hopkins University Baltimore, MD. http://www.ncbi.nlm.nih.gov/entrez/disomim.cgi2.id=304050
5. Fariello RG, Chun RW, Doro JM, Buncic JR, Prichard JS. EEG recognition of Aicardi's Syndrome. Arch Neurol 1977;34:563-6.
6. Weleber RG, Lovrien EW, Isom JB. Aicardi's syndrome: case report, clinical features and eletrophysiologic studies. Arch Ophthalmol 1978;96:285-90.
7. Hoyt CS, Billson F, Ouvrier R, Wise G. Ocular feature of Aicardi's syndrome. Arch Ophthalmol 1978;96:291-5.
8. Del Pero RA, Mets MB, Tripathi RC, Torczynski E. Anomalies of retinal architecture in Aicardi syndrome. Arch Ophthalmol 1986;104:1659-64.
9. Hashemi K, Traboulsi E, Chavis R, Scribanu N, Chrousos GA. Chorioretinal lacuna in the amniotic band syndrome. J Pediatr Ophthalmol Strabismus 1991;28:238-9.
10. Gurrieri F, Sammito V, Ricci B, Iossa M, Bellussi A, Nerig G. Possible new type of oral-facial-digital syndrome with retinal abnormalities: OFDS (type VIII) [commented on Am J Med Genet 1993;47:304-6]. Am J Med Genet, 1992;42:789-92.
11. Warburg M, Heuer HE. Autossomal dominant microcephaly with lacunar retinal hipopigmentations. In: 26º International Congress of Ophthalmology; 1983; Acta. Philadelphia: JB Lippincott; 1983. p.43-5.
12. Menezes AV, Lewis TL, Buncic JR. Role of ocular involvement in the prediction of visual development and clinical prognosis in Aicardi syndrome. Br J Ophthalmol 1996;80:805-11.
13. Hall-Crags MA, Harbord MG, Finn JP, Brett E, Kendall BE. Aicardi syndrome: MR assessment of brain structures and myelinization. AJNR Am J Neuroradiol1990;11:532-6.
14. Baierl P, Markl A, Thelen M, Laub MC. MR imaging in Aicardi syndrome. AJNR Am J Neuroradiol 1988;9:805-6.
15. Donnenfeld AE, Packer RJ, Zackai EH, Chee CM, Sellinger B, Emanuel BS. Clinical cytogenetic and pedigree findings in 18 cases of Aicardi syndrome. Am J Med Genet 1989;32:461-7.
16. Phillips HE, Carter AP, Kennedy JL, Rosman NP, O'Conner JF. Aicardi Syndromee: radiologic manifestations. Radiology 1978;127:453-5
Endereço para correspondência
Av. Angélica, 1757 5o Andar
São Paulo (SP) CEP 01227-200
Recebido para publicação em 28.11.2001
Aceito para publicação em 12.06.2002
Trabalho desenvolvido na Divisão de Oftalmologia da Faculdade de Medicina da Universidade de São Paulo (Hospital das Clínicas)
Nota Editorial: Pela análise deste trabalho e por sua anuência na divulgação desta nota, agradecemos aos Drs. Paulo Henrique de A. Morales e Juliana Maria Ferraz Sallum

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.