Arq. Bras. Oftalmol. 2001; 64 (6): 10.1590/S0004-27492001000600018
Total: 2884
Daniel Martins Moreira1; Juliano Soares Rabello Moreira1; Leandro Genehr Fritscher1; Régis Garcia de Garcia1; Samuel Rymer2
DOI: 10.1590/S0004-27492001000600018
RESUMO
Os anéis de Kayser-Fleischer (K-F) são alterações pigmentadas localizadas na membrana de Descemet, principalmente na região perilímbica na córnea. Estão associadas à doença de Wilson, sendo a manifestação oftalmológica mais comum da mesma e se correlacionam diretamente ao tempo de evolução desta doença. Os anéis de K-F caracterizam-se pela deposição de granulações de cobre de tamanhos e formas variadas na córnea e predominam na periferia corneana. A doença diretamente ligada ao aparecimento dos anéis de K-F, a doença de Wilson, caracteriza-se por distúrbio metabólico com acúmulo de cobre nos tecidos humanos, principalmente no fígado. A presença de anéis pigmentados na córnea nem sempre é diagnóstico de anéis de K-F, devendo ser diferenciados de outras alterações pigmentadas da córnea não ligadas à doença de Wilson. Estas englobam a vasta maioria de doenças hepato-biliares que podem ocasionar acúmulo de cobre, além de corpos estranhos intra-oculares contendo cobre, mieloma múltiplo, entre outras. O objetivo do presente artigo é revisar alguns aspectos desta alteração pigmentada corneana, os anéis de K-F, além de descrever algumas características da principal doença ligada ao seu aparecimento.
Descritores: Degeneração hepatolenticular; Doenças da córnea; Transtornos da pigmentação; Membrana de Descemet
ABSTRACT
Kayser-Fleischer (K-F) rings are characterized by copper deposition on Descemet's membrane, mainly near the limbus. They are strongly associated with Wilson's disease and are the most common ocular finding in this disease. There is an association between the presence of the rings and the time of disease. The K-F rings are composed of small copper granulations of different sizes on the cornea. Wilson's disease is characterized by an accumulation of copper in the body, primarily in the liver. The occurrence of pigmented rings on the cornea not associated with Wilson's disease have been reported and must be differentiated from the K-F rings. They occur most often in hepatobiliary diseases, multiple myeloma and ocular copper containing foreign bodies. The purpose of this paper is to review the K-F rings and Wilson's disease.
Keywords: Hepatolenticular degeneration; Corneal diseases; Pigmentation disorders; Descemet's membrane
INTRODUÇÃO
Os anéis pigmentados na córnea foram descritos primeiramente por Kayser, em 1902, em paciente com o suposto diagnóstico de esclerose múltipla. Em 1909, Fleischer relatou um caso de um paciente com anéis corneanos, cirrose hepática e pseudoesclerose. Atualmente, o termo "anéis de Kayser-Fleischer" (K-F) refere-se à coloração marrom-dourada, marrom-esverdeada, amarelo-esverdeada, amarelo-dourada ou bronze da membrana de Descemet na região perilímbica da córnea (Figura 1). É a manifestação oftalmológica mais comum da doença de Wilson (DW).
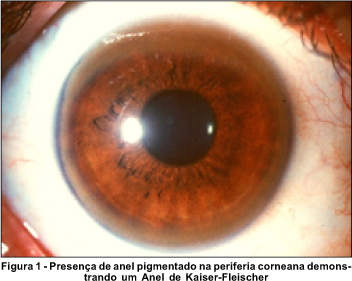
Freqüentemente, os anéis de K-F podem ser detectados a olho nu, especialmente em pacientes com íris de cor azulada ou esverdeada(1). O exame com lâmpada de fenda e a biomicroscopia, no entanto, são geralmente necessários para o diagnóstico definitivo. Os anéis são, caracteristicamente, mais pronunciados na periferia, ou seja, na região perilímbica e tendem a se tornar menos evidentes à medida que se aproximam da região central da córnea. São geralmente bilaterais e simétricos, mas existem relatos de anéis unilaterais(2-3).
Estudos utilizando microscopia eletrônica e histoquímica(4) mostraram que os anéis de K-F são compostos por densas e não uniformes camadas contendo granulações de cobre com tamanhos variados, separadas por intervalos sem deposições com diâmetros também variáveis, na membrana de Descement da córnea(5). Estes depósitos, entretanto, não representam a maior parte do cobre presente na córnea em indivíduos com doença de Wilson. Nesses casos, a maior deposição de cobre encontra-se na camada do estroma, como foi demonstrado em estudo utilizando histoquímica(4,6). Todavia, nessa camada não há formação de agregados pigmentares como ocorre na de Descemet. A concentração de cobre parece não ser maior na periferia em comparação à região central. A região periférica é a área onde ocorre a maior associação do cobre com compostos sulfatados, ligação considerada responsável pela produção dos anéis(4). As variações de cores entre os pacientes com anéis de K-F ou mesmo num único paciente ao longo do tempo ocorrem devido ao diferente número de granulações de cobre, tamanhos variados, posições diversas, concentrações variáveis. As variações de tamanho dos anéis de K-F estão correlacionadas com o tempo de doença. Estas variações parecem não estar muito relacionadas com a quantidade total de cobre no fígado e a quantidade excretada na urina, bem como com o nível de ceruloplamina sérica(7).
Dividem-se os anéis de K-F em quatro estágios conforme o grau de acometimento corneano: 0 - sem anel corneano identificável; 1 - anel no pólo superior apenas; 2 - anéis nos pólos superiores e inferiores; 3 - anel por toda a circunferência da córnea.
DOENÇA DE WILSON
A DW foi descrita em 1912 por S. A. Kinnier Wilson como uma degeneração hepatolenticular progressiva(8). Wilson descreveu uma síndrome familiar caracterizada por cirrose e degeneração lenticular. Atualmente, sabe-se que a DW é uma anormalidade do metabolismo, caracterizada pelo acúmulo de cobre nos tecidos humanos(9).
É uma doença hereditária com modo de transmissão autossômico recessivo e o gene responsável por esta anormalidade se encontra no cromossomo 13. Estima-se que a prevalência de indivíduos homozigóticos com o gene da doença, seja de aproximadamente 30 por milhão de pessoas, e a prevalência de heterozigotos seja de 90 por milhão, sendo assim considerada uma das doenças genéticas autossômicas recessivas mais prevalentes. Os heterozigotos não desenvolvem DW e tampouco necessitam de tratamento, podendo, no entanto, apresentar discretas anormalidades no metabolismo do cobre causando, às vezes, confusões diagnósticas(10). Diferentemente dos outros erros inatos do metabolismo, na DW o tratamento médico pode reverter dramaticamente o curso da deteriorização progressiva(11).
O defeito no metabolismo em pacientes com DW está na inabilidade do organismo de manter o balanço de cobre em torno de zero. O cobre presente na dieta normal é geralmente excessivo para as necessidades humanas. Cerca de metade do cobre ingerido na dieta é absorvido pelo trato gastrointestinal. Em indivíduos normais, o excesso de cobre é excretado pelo rim e pelo fígado. Nos pacientes com DW há acúmulo desse cobre no fígado(12). Ainda não se estabeleceu exatamente a causa desse acúmulo, assunto que ainda é alvo de vários estudos(12-14).
As manifestações clínicas relacionam-se com o depósito progressivo e gradual de cobre nos tecidos. A doença ocorre predominantemente em crianças, adolescentes e adultos jovens. Em crianças as manifestações hepáticas predominam, enquanto nos adolescentes e adultos observa-se sobretudo as manifestações neuropsiquiátricas. As manifestações hepáticas englobam um espectro amplo de doenças agudas e crônicas. Mais comumente têm curso crônico com características de cirrose pós-necrótica, insuficiência hepática progressiva, hipertensão porta, esplenomegalia, varizes gastroesofágicas e ascite. A peritonite bacteriana espontânea pode ocorrer. Por outro lado, a DW pode apresentar-se como um episódio de hepatite aguda, que usualmente regride espontaneamente e que pode equivocadamente ser atribuída à infecção viral.
Geralmente os pacientes irão apresentar manifestações neurológicas na segunda ou terceira década de vida. Estas manifestações são invariavelmente acompanhadas por anéis de K-F. Há relatos de pacientes com manifestações neuropsiquiátricas sem a presença de anéis de K-F(15-16). Estes, no entanto, são considerados exceções. O tamanho dos anéis de K-F está fortemente correlacionado com a severidade da doença(7).
Outra manifestação oftalmológica associada à DW é a catarata descrita como semelhante ao girassol, relatada em 1922 por Siemerling e Oloff. É uma alteração que não interfere com a visão. A catarata consiste em um anel central na região pupilar do cristalino com cor e extensões em forma de pétalas que se assemelham ao girassol. O cristalino apresenta depósitos granulares semelhantes ao da córnea(5,17).
As manifestações neurológicas iniciais incluem tremores, disartria, sialorréia, incordenação e ataxia. Os sintomas típicos, geralmente de início insidioso, caracterizam-se por tremor, distonia, rigidez, disartria, disfagia e alteração de marcha. O tremor, unilateral ou bilateral, é o sintoma mais freqüente e pode ser contínuo ou intermitente. Disartria é um achado precoce. Alterações psiquiátricas são freqüentes em pacientes com DW. Cerca de 20% dos pacientes apresentam anormalidades psiquiátricas como sintoma inicial da doença. Os achados psiquiátricos relacionados são: manifestações esquizofreniformes ou paranóides, distúrbios do humor, alucinações e ilusões, comportamento desinibido, perda do insight. Raramente os pacientes com DW terão demência. Esta, entretanto, é uma das poucas reversíveis e a sua característica é predominantemente subcortical(18).
Podem ocorrer alterações hematológicas como a hemólise intravascular, com teste de Coombs direto negativo. Freqüentemente a hemólise severa ocorre no início de uma hepatite fulminante. Problemas renais podem ocorrer sob a forma de acidose tubular renal distal ou proximal. Alterações reumatológicas de diversas formas também podem ser encontradas. Hiperpigmentação da pele é um achado dermatológico raro, assim como insuficiência cardíaca congestiva e arritmias. Manifestações endocrinológicas englobam ginecomastia e atraso na puberdade, e são devidos à disfunção hepática(6,10-11,19).
Na vasta maioria de pacientes com a doença, a presença de ambos anéis de K-F e hipoceruloplasminemia confirmam o diagnóstico. Os anéis, apesar de invariavelmente estarem presentes quando há manifestações neurológicas, muitas vezes estão ausentes em pacientes que apresentam apenas comprometimento hepático. Os valores normais para ceruloplasmina estão entre 20 e 40 mg/dL e são determinadas por técnicas enzimáticas ou imunocitoquímicas. Apesar da grande maioria dos doentes apresentar níveis abaixo de 20 mg/dL, alguns permanecem com valores entre 20 e 30 mg/dL(15,20-21). Um nível abaixo de 20 mg/dL, porém, por si só não é diagnóstico da doença(22), pois pode ser encontrado em 10 a 20% dos heterozigotos, em pacientes com hepatites fulminantes de qualquer etiologia, em 25% de crianças com hepatite crônica agudizada, neonatos normais, pacientes com malabsorção intestinal, nefrose ou malnutrição. Valores acima de 30 mg/dL praticamente excluem a doença(10).
Quando os anéis estão ausentes, hipoceruloplasminemia em conjunto com concentração hepática de cobre maior que 250 microgramas por grama de peso seco é suficiente para o diagnóstico. Essa medida é feita através de biópsia, que deve ser realizada com cuidados para que o material de coleta esteja livre de cobre(11). Se a hipoceruloplasminemia está ausente, a presença de anéis de K-F em conjunto com a concentração de cobre hepático maior que 250 microgramas por grama de peso é geralmente confirmatória.
Todos os pacientes homozigotos, sintomáticos ou não, requerem tratamento por toda a vida. Com o tratamento adequado pacientes assintomáticos assim permanecem e a grande maioria de pacientes sintomáticos tem melhora ou resolução dos seus sintomas.
Penicilamina permanece como primeira opção para o tratamento. Provavelmente produz seu efeito removendo o cobre dos tecidos em resultado de uma cupriurese dramática. A dose inicial está entre 1 a 2 gramas por dia divididas em 4 doses, via oral, meia hora antes das refeições ou 2 horas após as mesmas. Pacientes criticamente doentes podem receber inicialmente 3 a 4 gramas por dia. A dose pode também ser reduzida, à medida que os sintomas melhoram e que a excreção urinária de cobre decresça, para 0,5 a 1 grama por dia. Pacientes devem também receber piridoxina 25 mg por dia devido ao efeito antipiridoxina da medicação. A penicilamina, entretanto, é uma droga com vários e importantes efeitos adversos que muitas vezes limita o seu uso(10).
Trientina é um agente quelante alternativo que pode ser usado em pacientes ocasionais que são intolerantes à penicilamina e para decuperização inicial. Parece ser útil em prevenir a deteriorização clínica rápida muitas vezes vistas em pacientes que descontinuaram terapia com penicilamina. É administrada via oral em doses de 750 a 2000 mg diariamente divididas em 3 doses(10).
Zinco oral é atualmente considerado como uma terceira opção, sendo utilizado apenas em pacientes intolerantes a ambas as medicações anteriores. Dieta pobre em cobre não consegue alcançar um balanço negativo de cobre. Recomenda-se, porém, que no início do tratamento evite-se alimentos com muito cobre como frutos do mar, fígado, chocolates e cogumelos(10,23-24).
Finalmente, o tratamento definitivo para a doença pode ser o transplante hepático. É uma opção terapêutica interessante pois corrige o erro do metabolismo e, dessa forma, acaba com a necessidade de tratamento médico contínuo.
OUTRAS DOENÇAS
Anéis pigmentares de córnea também foram descritos em outras doenças além da DW e, portanto, devem ser diferenciados dos anéis de K-F. Pseudoanéis de K-F já foram associados à cirrose biliar primária(11,17,25). Nessa doença, devido à existência de colangite destrutiva com redução de saída de bile e ao fato de que o excesso de cobre é excretado pela via biliar, há acúmulo de cobre no organismo. Este, pode ser verificado pela deposição desse elemento nos tecidos, como na córnea, por exemplo. Estes anéis, entretanto, podem ser diferenciados dos anéis da DW pois são menos densos e possuem cor mais bronzeada do que marrom(6). Da mesma forma, pacientes com marcada colestase por outras etiologias, hepatopatia alcoólica crônica(26), hepatite autoimune crônica(27), cirrose criptogênica(28), cirrose colestática familiar(29), atresia de vias biliares(30), podem apresentar anéis pigmentares muito semelhantes aos anéis de K-F.
Corpos estranhos intraoculares contendo cobre em concentrações menores que 85% estão associados à formação de anéis por deposição de cobre que lentamente se difunde até o cristalino e a córnea. Corpos estranhos com concentrações de cobre superiores a 85% geralmente causam uma forte reação inflamatória no olho com conseqüências catastróficas(6).
Hipercarotenemia associada ao arco senil prévio foi observado e considerado como diagnóstico diferencial de anéis de K-F(31). Da mesma forma, arcos pigmentares de córnea já foram observados em infestação por Schistosoma japonicum, tripanossomose africana e uso tópico prolongado de cobre para tratamento de tracoma ou outra doença ocular(6). Mieloma múltiplo com altos níveis de gamaglobulinas e cobre também pode se apresentar com depósitos corneanos semelhantes aos anéis de K-F. Diferem, no entanto, por estarem geralmente localizados na região central da córnea.
TRATAMENTO
Ainda há muito a se investigar sobre o comportamento dos anéis de K-F com o tratamento da DW. Há relatos de redução dos anéis de K-F com o uso de penicilamina. Sabe-se que cerca de 90% dos pacientes terão melhora com o tratamento com penicilamina em um ano(6). O desaparecimento completo dos anéis de K-F com tratamento com penicilamina já foi descrito, porém é consenso, na literatura, como algo bastante raro(23).
O tratamento com zinco oral, uma das opções de tratamento para a DW, mostrou sensíveis reduções no diâmetro dos anéis de córnea. A média de redução foi de 14% por ano de tratamento. Esta regressão, no entanto, não se correlacionam com a melhora dos sintomas, especialmente os neurológicos(23).
O uso do dimercaprol, em um estudo de pequeno porte, mostrou redução da quantidade corneana de cobre em até 45% sem, no entanto, reduzir o tamanho dos anéis(32).
Pacientes com DW que foram submetidos a transplante hepático geralmente apresentam sensíveis reduções das anormalidades corneanas, muitas vezes, até o completo desaparecimento dos anéis de K-F. Num estudo com quatro pacientes(33), o desaparecimento completo ocorreu em um período de oito meses até cinco anos, dependendo da extensão dos anéis. Em três meses, já se pode observar alguma mudança em relação ao estado basal.
REFERÊNCIAS
1. Cairns JE, Williams HP, Walshe JM. "Sunflower cataract" in Wilson's disease. Brit Med J 1969;3:95-6.
2. Innes JR, Strachan IM, Triger DR. Unilateral Kayser-Fleischer ring. Br J Ophthalmol 1986;70:469-70.
3. Madden JW, Ironside JW, Triger DR, Bradshaw JP. An unusual case of Wilson's disease. Q J Med 1985;55:63-73.
4. Johnson RE, Campbell RJ. Wilson's disease. Electron microscopic, x-ray energy spectroscopic, and atomic absorption spectroscopic studies of corneal copper deposition and distribution. Lab Invest 1982;46:564-9.
5. Tso MOM, Fine BS, Thorpe HE. Kayser-Fleischer ring and associated cataract in Wilson's disease. Am J Ophthalmol 1975;79:479-88.
6. Wiebers DO, Hollenhorst RW, Goldstein NP. The ophthalmologic manifestations of Wilson's disease. Mayo Clinic Proc 1977;52:409-16.
7. Rodman R, Burnstine M, Esmaeli B, Sugar A, Martonyi C, Johnson V, et al. Wilson's disease presynmptomatic patients and Kayser-Fleischer rings. . Ophthalmic Genet 1997; 18:79-85.
8. Wilson SAK. Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver. Brain 1912;34:295-509.
9. Herron BE. Wilson's disease (hepatolenticular degeneration). Ophthalmic Semin 1976;1:63-9.
10. Yarze JC, Martin P, Muñoz SJ, Friedman LS. Wilson's disease: current status. Am J Med 1992;92:643-4.
11. Tauber J, Steinert RF. Pseudo-Kayser-Fleischer ring of the cornea associated with non-Wilsonian liver disease. A case report and literature review. Cornea 1993;12:74-7.
12. Ferenci P. Wilson's disease. Ital J Gastroenterol Hepatol 1999;31:416-25.
13. Pfeil SA, Lynn DJ. Wilson's disease: copper unfettered. J Clin Gastroenterol 1999;29:22-31.
14. Bacon BR, Schilsky ML. New knowledge of genetic pathogenesis of hemochromatosis and Wilson's disease. Adv Intern Med 1999;44:91-116.
15. Ross ME, Jacobson IM, Dienstag JL, Martin JB. Late-onset Wilson's disease with neurological involvement in the absence of Kayser-Fleischer rings. Ann Neurol 1985;17:411-3.
16. Demirkiran M, Jankovic J, Lewis RA, Cox DW. Neurologic presentation of Wilson's disease without Kayser-Fleischer rings. Neurology 1996;46:1040-3.
17. Fleming CR, Dickson ER, Hollenhorst RW, Goldstein NP, McCall JT, Baggenstoss AH. Pigmented corneal rings in a patient with primary biliary cirrhosis. Gastroenterology 1975;69:220-5.
18. LeWitt PA, Pfeiffer RF. Neurologic aspects of Wilson´s disease: clinical manifestations and treatment considerations In: Jankovic J, Tolosa E, eds. Parkinson's disease and movement disorders. Baltimore: Urban & Schwarzenberg; 1998. p.377-400.
19. Brodland DG, Bartley GB. Kayser-Fleischer rings in a patient with basal cell carcinoma: fortuitous diagnosis of presymptomatic Wilson's disease. Mayo Clin Proc 1992;67:142-143.
20. Yüce A, Kocak N, Ozen H, Gürakan F. Wilson's disease patients with normal ceruloplasmin levels. Turk J Pediatr 1999;41:99-102.
21. Cauza E, Maier-Dobersberger T, Polli C, Kaserer K, Kramer L, Ferenci P. Screening for Wilson's disease in patients with liver diseases by serum ceruloplasmin. J Hepatol 1997;27:358-62.
22. Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, et al. Wilson's disease in patients presenting with liver disease: a diagnostic challenge. . Gastroenterology 1997;113:212-8.
23. Esmaeli B, Burnstine MA, Martonyi CL, Sugar A, Johnson V, Brewer GJ. Regression of Kayser-Fleischer rings during oral zinc therapy: correlation with systemic manifestations of Wilson's disease. Cornea 1996;15:582-8.
24. Brewer GJ. Practical recommendations and new therapies for Wilson's disease. Drugs 1995;50:240-9.
25. Fleming CR, Dickson ER, Wahner HW, Hollenhorst RW, McCall JT. Pigmented corneal rings in non-Wilsonian liver disease. Ann Intern Med 1977;86:285-8.
26. Weinberg LM, Brasitus TA, Lefkowitch JH. Fluctuating Kayser-Fleischer-like rings in a jaundiced patient. Arch Intern Med 1981;141:246-247.
27. Zargar SA, Thapa BR, Sahni A, Mehta S. Kayser-Fleischer like ring in autoimmune chronic active hepatitis. Indian J Gastroenterol 1991;10:101-2.
28. Rimola A, Bruguera M, Rodés J. Kayser-Fleischer-like rings in a cryptogenic cirrhosis. Arch Intern Med 1978;138:1857-8.
29. Kaplinsky C, Sternlieb I, Javitt N, Rotem Y. Familial cholestatic cirrhosis associated with Kayser-Fleischer rings. Pediatrics 1980;65:782-8.
30. Frommer D, Morris J, Sherlock S, Abrams J, Newman S. Kayser-Fleischer-like rings in patients without Wilson's disease. Gastroenterology 1977;72: 1331-5.
31. Giorgio AJ, Cartwright GE, Wintrobe MM. Pseudo-Kayser-Fleischer rings. Arch Intern Med 1964;113:817-8.
32. Belkin M, Zeimer R, Chajek T, Friedman G, Melamed E. Non-invasive quantitation of corneal copper in hepatolenticular degeneration (Wilson's disease). Lancet 1976;1:391-2.
33. Song HS, Ku WC, Chen CL. Disappearance of Kayser-Fleischer rings following liver transplantation. Transplant Proc 1992;24:1483-5.
Serviço de Oftalmologia do Hospital de Clínicas de Porto Alegre. Faculdade de Medicina da Universidade Federal do Rio Grande do Sul.
1Doutorandos da Faculdade de Medicina da Universidade Federal do Rio Grande do Sul.
2 Professor Adjunto do Departamento de Oftalmologia da Universidade Federal do Rio Grande do Sul.
Endereço para correspondência: Rua Machado de Assis, 673 - Porto Alegre (RS) CEP 90620-260. E-mail: [email protected]

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.