Arq. Bras. Oftalmol. 2013; 76 (2): 10.1590/S0004-27492013000200015
Total: 1694
Paolo Rama1; Karl Anders Knutsson1; Carmen Rojo2; Paola Carrera3; Maurizio Ferrari3
DOI: 10.1590/S0004-27492013000200015
ABSTRACT
We report an atypical case of granular corneal dystrophy recurrence after deep anterior lamellar keratoplasty. We describe clinical features, histopathological analysis of the lamellar graft specimen and DNA analysis results. The slit-lamp examination and histopathological findings from the graft specimen indicated the confinement of the typical deposits of granular corneal dystrophy deep in the graft interface area. This localization is atypical, since in most cases recurrences in grafts tend to be initially superficial and situated in the epithelial or subepithelial corneal layers. Molecular genetic analysis revealed an already described mutation and a new intronic variant. The unusual localization and timing of this recurrence of granular corneal dystrophy after deep anterior lamellar keratoplasty suggests that corneal stromal keratocytes may play a role in the formation of granular deposits.
Keywords: Corneal transplantation; Corneal dystrophies, hereditary; Cornea; Corneal stroma; Corneal keratocytes; Epithelium, corneal; Recurrence
RESUMO
É relatado um caso atípico de recorrência de distrofia corneana granular após transplante lamelar anterior profundo. São descritas as características clínicas, a análise histopatológica do espécime do enxerto lamelar e os resultados de análises de DNA. O exame com lâmpada de fenda e a análise histopatológica do espécime do enxerto demonstram o confinamento dos depósitos típicos da distrofia corneana granular profundamente, na área de interface do enxerto. Esta localização é atípica, uma vez que, na maioria dos casos de recidivas em enxertos, estes tendem a ser no início localizados superficialmente, nas camadas epiteliais ou subepitelial da córnea. A análise genética molecular revelou uma mutação já descrita e uma nova variante intrónica. A localização incomum e o tempo de aparecimento da presente recorrência da distrofia corneana granular após transplante lamelar anterior profundo sugere que ceratócitos do estroma corneano possam desempenhar algum papel na formação dos depósitos granulares.
Descritores: Transplante de córnea; Distrofias hereditárias da córnea; Córnea; Substância própria; Ceratócitos da córnea; Epitélio anterior; Recidiva
CASE REPORTS RELATOS DE CASOS
Unusual early recurrence of granular dystrophy after deep anterior lamellar keratoplasty: case report
Recorrência atípica e precoce de distrofia granular após transplante lamelar profundo: relato de caso
Paolo RamaI; Karl Anders KnutssonI,II; Carmen RojoII; Paola CarreraIII,IV; Maurizio FerrariIII,IV,V
IOphthalmology - Cornea and Ocular Surface Unit, San Raffaele Scientific institute, Milano
IIDepartment of Ophthalmology, San Raffaele Scientific Institute, Milano
IIIGenomic Unit for the Diagnosis of Human Pathologies, Center for Genomics, Bioinformatics and Biostatistics, San Raffaele Scientific Institute, Milano
IVDiagnostica e Ricerca San Raffaele SpA, Milano
VVita-Salute San Raffaele University, Milano
ABSTRACT
We report an atypical case of granular corneal dystrophy recurrence after deep anterior lamellar keratoplasty. We describe clinical features, histopathological analysis of the lamellar graft specimen and DNA analysis results. The slit-lamp examination and histopathological findings from the graft specimen indicated the confinement of the typical deposits of granular corneal dystrophy deep in the graft interface area. This localization is atypical, since in most cases recurrences in grafts tend to be initially superficial and situated in the epithelial or subepithelial corneal layers. Molecular genetic analysis revealed an already described mutation and a new intronic variant. The unusual localization and timing of this recurrence of granular corneal dystrophy after deep anterior lamellar keratoplasty suggests that corneal stromal keratocytes may play a role in the formation of granular deposits.
Keywords: Corneal transplantation; Corneal dystrophies, hereditary; Cornea/pathology; Corneal stroma; Corneal keratocytes; Epithelium, corneal/cytology; Recurrence
RESUMO
É relatado um caso atípico de recorrência de distrofia corneana granular após transplante lamelar anterior profundo. São descritas as características clínicas, a análise histopatológica do espécime do enxerto lamelar e os resultados de análises de DNA. O exame com lâmpada de fenda e a análise histopatológica do espécime do enxerto demonstram o confinamento dos depósitos típicos da distrofia corneana granular profundamente, na área de interface do enxerto. Esta localização é atípica, uma vez que, na maioria dos casos de recidivas em enxertos, estes tendem a ser no início localizados superficialmente, nas camadas epiteliais ou subepitelial da córnea. A análise genética molecular revelou uma mutação já descrita e uma nova variante intrónica. A localização incomum e o tempo de aparecimento da presente recorrência da distrofia corneana granular após transplante lamelar anterior profundo sugere que ceratócitos do estroma corneano possam desempenhar algum papel na formação dos depósitos granulares.
Descritores: Transplante de córnea/efeitos adversos; Distrofias hereditárias da córnea; Córnea/patologia; Substância própria; Ceratócitos da córnea; Epitélio anterior/citologia; Recidiva
INTRODUCTION
Granular corneal dystrophy (GCD) is a bilateral autosomal dominant corneal dystrophy characterized by small, breadcrumb-like, grayish-white, stromal opacities(1). In early stages, lesions occupy the anterior central stroma, but later can coalesce and occupy deeper stromal layers(2).Visual function usually remains good until the fifth decade and most patients require no treatment as visual acuity remains adequate for their needs. However, if opacities become denser and occupy the visual axis, corneal transplantation may be necessary. Most authors agree that typical recurrences are superficial and early involvement is noted to be central and epithelial(2). Later on, the recurrent material adopts the classic breadcrumb-like appearance of GCD, gradually involving subepithelial and stromal layers. Lyons et al.(2), reported that the time of recurrence can range from 13 to 73 months and shows no significant difference between penetrating keratoplasty (PK) and lamellar keratoplasty (LK). This case report describes the clinical, histopathological and molecular genetic findings of an atypical case of GCD recurrence.
METHODS
The corneal specimen was stained with hematoxylin and eosin, Congo red, Masson trichrome and periodic acid-Schiff (PAS). After pre-test genetic counseling, the patient gave her consent to the molecular analysis. Genomic DNA was extracted from a peripheral blood leukocyte sample using standard protocols. Transforming growth factor beta-induced gene (TGFBI also known as BIGH3) whole coding region and exon junctions were analyzed by directly sequencing specific polymerase chain reaction (PCR) products on both strands. Dye terminator reaction sequences were loaded on a 3730AB (Applied Biosystems, Foster City, CA) automatic sequencer. Sequences were assembled and compared to the reference ENSG00000120708 with the Sequencer V.4 (GeneCodes Co., Ann Arbor, MI).
RESULTS
Clinical findings
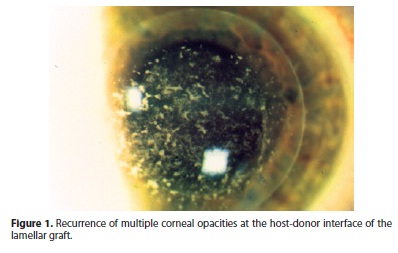
A 43-year-old Caucasian female was diagnosed with advanced GCD. Best spectacle-corrected visual acuity (BSCVA) was 0.2 (20/100) for the right eye and 0.5 (20/40) for the left eye. In January 2000, she underwent deep anterior lamellar keratoplasty (DALK) on the right eye, with no complications. During the six month follow-up, BSCVA was 0.63 (20/30) and on slit-lamp examination she presented an early recurrence of corneal opacities in the host-donor interface (Figure 1). Progressive worsening of the disease required PK in March 2001 on the same eye. After two years, minor recurrence developed, initially involving the subepithelial corneal layers and later the superficial stroma. In October 2005, PK was performed on the left eye and minor epithelial recurrence was observed after a period of two years.
Histology report
Deep stromal deposits were observed at the host-donor interface. They were eosinophilic in tissue samples stained with hematoxylin and eosin and had a bright red color in samples stained with Masson trichrome (Figure 2). Sections stained with PAS and Congo red were negative and the granules lacked birefringence on polarization.

Molecular genetic analysis
Sequence analysis revealed the presence of TGFBI heterozygous variants: i) the c.1663C>T transition resulting in the p.Arg555Trp missense substitution in the putative protein, already described in association with corneal dystrophy, particularly the GCD type 1 (Groenouw's type 1 granular corneal dystrophy) (1); ii) a novel intronic variant IVS16-5T>C; iii) a number of known variants reported in dbSNP as polymorphisms (IVS6+119A>G, IVS7+47T>C, c.981A>G, IVS10-43A>G, c.1417C>T, IVS11-252T>C, c.1620T>C, IVS12+23G>A, IVS13-71A>G, IVS13-54T>A, IVS14+44T>C, IVS15-73G>A). The patient reported paternal-side family history; however, we could not analyze her aged parents.
DISCUSSION
Mutations in the TGFBI gene on chromosome 5q31 are associated with phenotypically distinct corneal dystrophies, including GCD(1).The transforming growth factor-beta induced protein, also known as keratoepithelin (KE), is an adhesion protein which is strongly expressed by the corneal epithelium, by other corneal cells in minor quantities and is a component of the extracellular matrix in many tissues. It is secreted by corneal epithelial cells and can be found in normal stroma, covalently bound to type VI collagen. KE is a small molecule, about the size of albumin, which allows diffusion from its epithelial source to the stroma.
Cases of deep stromal involvement in suture canals and the host-donor interface have been reported(3,4), but are less common than superficial recurrences(2). In our case, the opacities were atypical as they recurred after a short period of time (6 months) and occurred only in the deep layers of the host-donor interface. Numerous mechanisms could explain this particular case. Mutated KE secreted by epithelial cells can diffuse across the graft-host junction and localize in the deeper stromal layers at the interface. Alteration of the host keratocytes might be responsible for the localization of the deposits in the stroma. Alternatively, host keratocytes could be directly responsible for the deposition of the material. Production of abnormal material has been reported after LASIK in patients with Avellino corneal dystrophy(5) and radial keratectomy in a patient with GCD(6). In these cases, collagen damage may determine keratocyte activation, favoring the production of deposits, and an identical mechanism could be hypothesized for eyes that undergo DALK surgery.
In our patient, molecular analysis revealed the presence of several variants, mostly common polymorphisms; in addition the p.Arg555Trp mutation and a new intronic unclassified variant (IVS16-5T>C) were detected. The p.Arg555Trp has been reported several times and noticeably, if compared to other TGFBI mutations, it has been correlated with a lower severity(7) as assessed by age at first graft and time to recurrence. On the contrary, a higher degree of severity has been described in patients homozygous for the p.Arg555Trp. In our patient, we observed a rather severe phenotype, characterized by early onset and multiple recurrences; we would argue that the IVS16-5T>C, located near the splice acceptor site of intron 16 might possibly have a negative influence on the splicing process.
Great advances have been made in the characterization of stromal corneal dystrophies, especially after the discovery of various mutations in the TGFBI gene and the different phenotypic alterations that can arise. Almost all studies point to an epithelial origin of the deposits, as KE is indeed produced by these cells. However, no studies have been able to explain how KE deposits accumulate in the stromal layers. Our case is unique because the deposits localize at the host-donor interface over a short period of time, suggesting that the host keratocytes might play a role in the formation of the characteristic deposits.
ACKNOWLEDGEMENTS
Special thanks to Dr. Pietro Maria Donisi of the Department of Pathology, General Hospital of Venice, Italy.
REFERENCES
1. Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7.
2. Lyons CJ, McCartney AC, Kirkness CM, Ficker LA, Steele AD, Rice AS. Granular corneal dystrophy. Visual results and pattern of recurrence after lamellar or penetrating keratoplasty. Ophthalmology. 1994;101(11):1812-7.
3. Salouti R, Hosseini H, Eghtedari M, Khalili MR. Deep anterior lamellar keratoplasty with Melles technique for granular corneal dystrophy. Cornea. 2009; 28(2):140-3.
4. Frising M, Wildhardt G, Frisch L, Pitz S. Recurrent granular dystrophy of the cornea: an unusual case. Cornea 2006; 25: 614-7.
5. Roh MI, Grossniklaus HE, Chung SH, Kang SJ, Kim WC, Kim EK. Avellino corneal dystrophy exacerbated after LASIK: scanning electron microscopic findings. Cornea. 2006;25(3):306-11.
6. Feizi S, Pakravan M, Baradaran-Rafiee AR, Yazdani S. Granular corneal dystrophy manifesting after radial keratotomy. Cornea. 2007;26(10):1267-9.
7. Ellies P, Renard G, Valleix S, Boelle PY, Dighiero P. Clinical outcome of eight BIGH3-linked corneal dystrophies. Ophthalmology. 2002;109(4):793-7.
 Corresponding address:
Corresponding address:
Rama Paolo.
Via Olgettina 60. San Raffaele Hospital
Cornea and Ocular Surface Unit - Milano, Italy
E-mail: [email protected]
Submitted for publication: February 1, 2013
Accepted for publication: May 8, 2013
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: P.Rama, None; K.A.Knutsson, None; C.Rojo, None; P.Carrera, None; M.Ferrari, None.
The study was performed with informed consent and following all the guidelines for experimental investigations required by the Institutional Review Board or Ethics Committee of which all authors are affiliated.
Study carried out at San Raffaele University, Milano, Italy.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.