INTRODUCTION
Sweet syndrome (acute febrile neutrophilic dermatosis) is characterized by fever, neutrophilic leukocytosis, and abrupt appearance of painful erythematous nodules and plaques, particularly on the face, neck, and limbs( 1 ). The most frequent ocular manifestations include conjunctivitis, episcleritis, inflammatory glaucoma, scleritis, iritis, and limbal nodules( 2 ). In this study, we report a very rare case of Sweet syndrome in which the patient presented nodular scleritis and peripheral ulcerative keratitis during the dermatologically inactive period of the disease. To the best of our knowledge, there is only one case of Sweet syndrome associated with peripheral keratitis in the literature, but during the dermatologically active period( 3 ).
CASE REPORT
46-year-old woman presented to a dermatologist with a 7-day history of painful redness on both arms and right hand, oral mucosal lesions that flared up and regressed during this time period, and arthralgia. Physical examination revealed red erythematous papules and vesicles across the right hand, between the fingers, and on the bilateral distal extensor areas of the upper extremities. There were no other systemic symptoms. Skin biopsy revealed neutrophilic inflammatory infiltrates in the dermis with no evidence of vasculitis (Figures 1-2). Laboratory tests revealed normal hemoglobin levels and platelet and leukocyte counts with neutrophilia [neutrophils: 8.29 × 109/L (normal: 2.06-7.02 × 109/L)]. The erythrocyte sedimentation rate (ESR) was elevated at 53 mm/h (normal: 0.00-20.00 mm/h) and C-reactive protein (CRP) levels were elevated at 1.06 mg/dL (normal: 0.00-0.50 mg/dL). The patient was referred to the otolaryngology, hematology, cardiology, rheumatology, and neurology departments for investigation of respiratory tract infection, hematologic malignancy, heart involvement, artralgia, and central nervous system involvement. She was hospitalized by the rheumatology department to investigate the etiology of arthralgia. She was further evaluated for systemic autoimmune diseases (with tests for antinuclear antibody, rheumatoid factor, antineutrophilic cytoplasmic antibody, and mitochondrial antibody) as well as for Brucella or hepatitis B infection, but all laboratory results were negative. The laboratory and clinical findings were consistent with Sweet's syndrome. Treatment was started with 32 mg/day oral methylprednisolone, which was tapered slowly over several weeks.
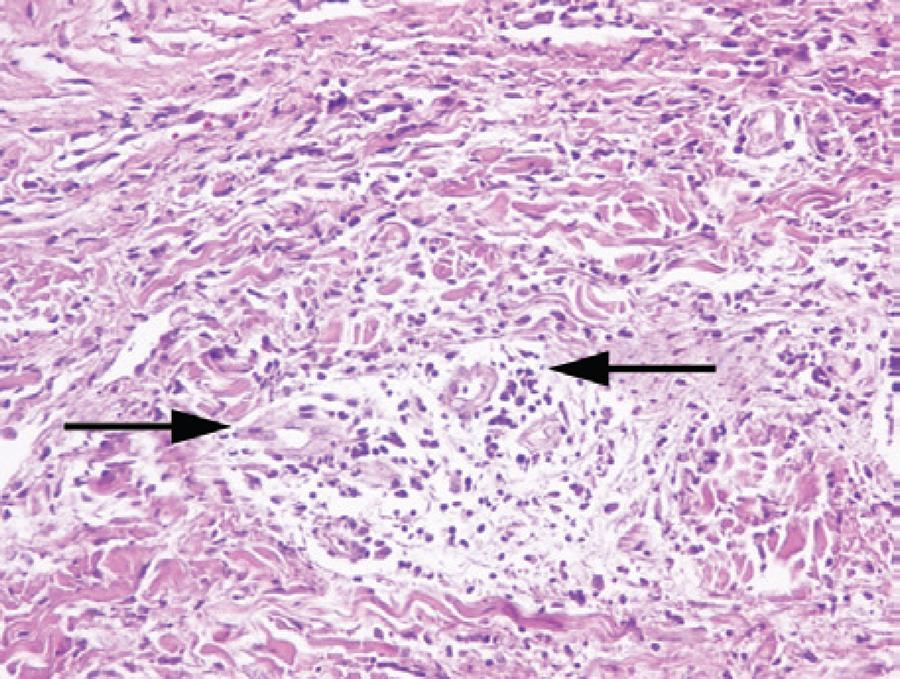
Figure 1 Perivascular mixed-type inflammatory infiltrate and edema without vasculitis (H&E stain, ×20).
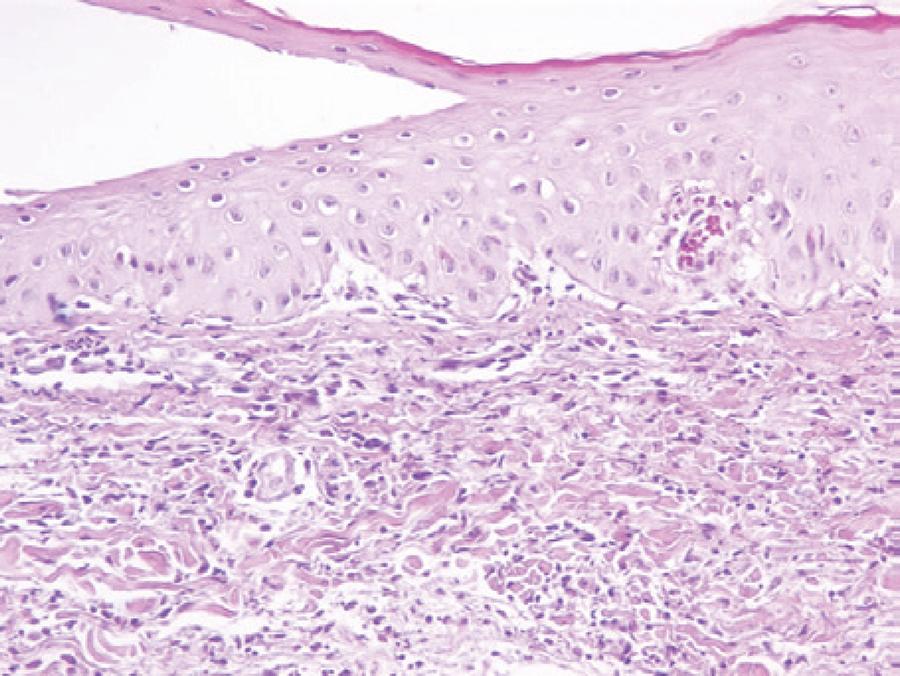
Figure 2 Neutrophilic infiltrate and edema in the upper dermis and exocytosis of neutrophils in the epidermis (H&E stain, ×20).
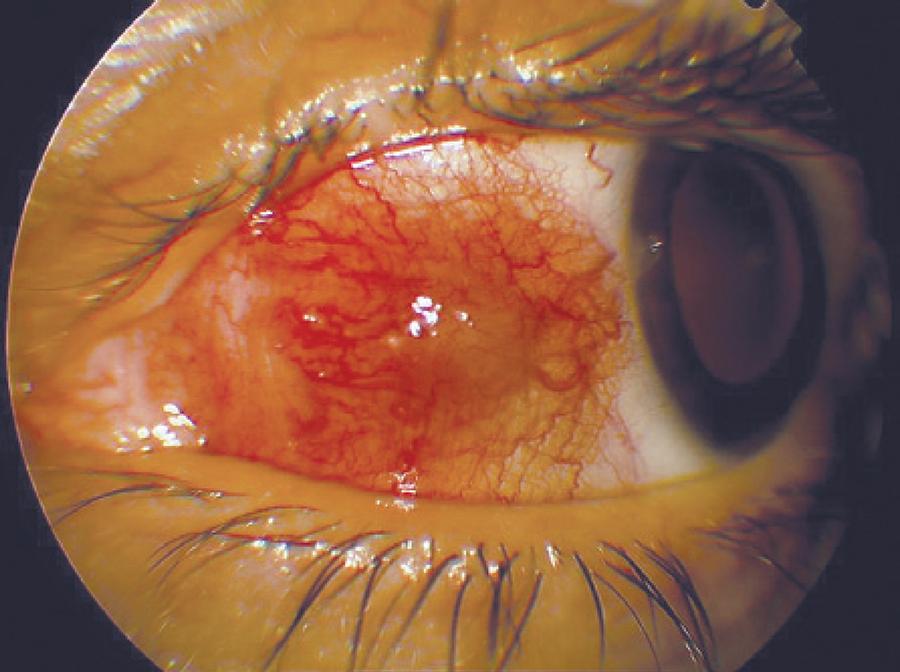
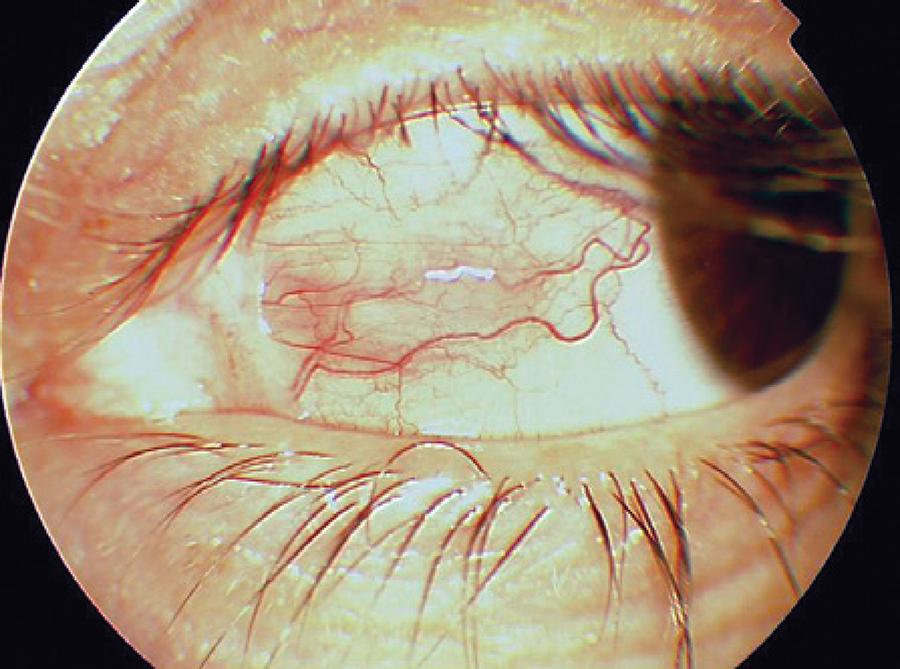
Five months later, the patient complained of a red and painful left eye that lasted for 10 days. She did not undergo any treatment for 2 months. Dermatological examination did not reveal any active lesions. Her pain was exacerbated with ocular movements. Ocular examination showed an uncorrected visual acuity of 10/10 in both eyes. Her intraocular pressure was 15 mmHg in the right eye and 16 mmHg in the left eye. An elevated and hyperemic scleral area (10 × 5 mm in size with surrounding episcleral injection) was observed on the nasal side of the left eye; peripheral ulcerative keratitis was observed adjacent to the nodular scleritis (Figure 3). There were no signs of anterior chamber inflammation. Posterior segment examination was unremarkable in the left eye and the nasal retina had mild pigment changes in the right eye. The patient was treated with fluorometholone 0.1% (Flarex, Alcon, USA), ofloxacin 0.3% (Okacin, Novartis, USA), and artificial tear eye drops 5 times a day in the left eye and 40 mg daily oral fluocortolone. Immediate improvement was observed, and within 2 days, nodular scleritis and peripheral keratitis had regressed. Seven days later, peripheral keratitis had healed and ofloxacin 0.3% was stopped (Figure 4). Two weeks after using 40 mg/ day oral fluocortolone and fluorometholone 0.1% eye drops, both medications were tapered slowly over several weeks and stopped.
DISCUSSION
Sweet syndrome is an uncommon, reactive dermatological disease with an unknown etiology. The features of the syndrome were first described by Dr. Robert Douglas Sweet in 1964( 4 ). Two of the major criteria and at least 2 of the 4 minor criteria are required to establish the diagnosis of Sweet’s syndrome (Table 1)( 1 , 5 ).
Table 1 Diagnostic criteria for Sweet syndrome
| Major criteria | Abrupt appearance of painful erythematous nodules and plaques. |
| Histopathological evidence of a dense dermal neutrophilic infiltrate without vasculitis. | |
| Minor criteria | Fever (greater than 38°C). |
| Response to treatment with systemic corticosteroids, potassium iodide, or colchicines. | |
| Association with an underlying hematological or visceral malignancy, inflammatory disease, or a previous upper respiratory or gastrointestinal infection, vaccination, or pregnancy. | |
| Abnormal laboratory values (erythrocyte sedimentation rate greater than 20 mm/h, elevated C-reactive protein, leukocytes greater than 8.0 × 109/L, and neutrophils greater than 70%). |
Sweet syndrome predominantly affects women and most frequently occurs between the ages of 30 to 50 years. However, children and younger adults may also be affected by the disease( 6 ). Patients may have extracutaneous manifestations involving the eyes, kidneys, bone, liver, central nervous system, intestines, heart, mouth, muscles, spleen, and lungs. Of all cases 15%-20% are associated with hematological malignancies (most commonly acute myelogenous leukemia) and solid tumors (most commonly carcinomas of the genitourinary organs, breast, and gastrointestinal tract). Ocular involvement has been reported in 4%-72% of patients with Sweet syndrome and ocular manifestations include conjunctivitis, episcleritis, subconjunctival hemorrhage, scleritis, inflammatory glaucoma, choroiditis, iritis, optic nerve involvement, limbal nodules, periorbital eruption, or orbital and eyelid inflammation with vesicular lesions( 5 , 7 , 8 ). Systemic corticosteroids (oral prednisone or prednisolone) are the mainstays of treatment for Sweet syndrome( 9 ). The ocular lesions of patients have resolved with systemic corticosteroids and topical corticosteroid eye drop treatment. Although the ocular lesions respond well to treatment, the dermatological lesions may persist or recur even when the patient is on systemic corticosteroid treatment( 2 ). Our case indicates that ulcerative keratitis and scleritis may be associated with active or inactive dermatological inflammatory diseases and should prompt systemic and laboratory investigation.

