Arq. Bras. Oftalmol. 2012; 75 (3): 10.1590/S0004-27492012000300012
Total: 4225
Maria Del Carmen Mendez; Yerena Muiños; Gonzalo Blanco; Maria Antonia Saornil; Ciro García-Alvarez; Jose Luis Sarasa1; Carmen Valbuena
DOI: 10.1590/S0004-27492012000300012
ABSTRACT
Rhabdomyosarcoma is a rare tumor, with an annual incidence of 4.3 cases per million children. Even thought, it is the most common soft tissue sarcoma in childhood, with a mean age of 6 to 8 years at diagnosis. A 4 year-old boy presented with a history of a fast growing (1-month) nodular lesion in the caruncle of his left eye. Slit lamp examination showed a vascularized solid nodular lesion in the semilunar fold. The lesion was surgically removed obtaining infiltrated edges with tumoral cells. A second surgery was performed with free tumour edges. The diagnosis of embryonal rhabdomyosarcoma, botryoid type, of intermediate differentiation was made. The treatment for botryoid rhabdomyosarcoma is basically surgical with the combined use of adjuvant polychemotherapy. In adolescent or adult patients (not in infants where growth bone disturbances can occur) external beam radiotherapy can be combined with chemotherapy. Rhabdomyosarcomas of the conjunctiva without orbital extension are rarely reported. We presented a case of a child with a rare tumor which we had a high suspicion of malignancy and early diagnosis and treatment and child is free of systemic disease 6 and half years later. Our research group believes that the key in these tumors is the high index of suspicion and early treatment.
Keywords: Rhabdomyosarcoma; Conjuctival neoplasms; Rhabdomyosarcoma, embryonal; Humans; Male; Children; Case report
RESUMO
Rabdomiossarcoma é um tumor raro, com uma incidência anual de 4,3 casos por milhão de crianças. É o sarcoma de partes moles mais comum na infância, com uma idade média de 6 a 8 anos no momento do diagnóstico. Um menino de 4 anos apresentou-se com uma história de um crescimento rápido (1 mês) lesão nodular em carúncula de seu olho esquerdo. O exame biomicroscópico mostrou uma lesão vascularizada sólida nodular na prega semilunar. A lesão foi removida cirurgicamente com obtenção de bordas infiltradas com células tumorais, segunda cirurgia foi feita com bordas tumorais livres. O diagnóstico de rabdomiossarcoma embrionário, tipo botrióide, de diferenciação intermediária foi feita. O tratamento para a rabdomiossarcoma botrióide é basicamente cirúrgico com o uso combinado de poliquimioterapia adjuvante. Em pacientes adolescentes e adultos (e não em lactentes onde os distúrbios do crescimento ósseo pode ocorrer) radioterapia externa pode ser combinada com a quimioterapia. Rabdomiossarcomas da conjuntiva sem extensão orbital são raramente relatadas. Apresentamos um caso de uma criança com um tumor raro que tivemos uma alta suspeita de malignidade e diagnóstico precoce e tratamento e criança é livre de doença sistêmica 6 anos e meio depois. Nosso grupo de pesquisa acredita que a chave para esses tumores é o alto índice de suspeita e tratamento precoce.
Descritores: Rabdomiossarcoma; Neoplasias da tunica conjuntiva; Rabdomiossarcoma embrionário; Humanos; Masculino; Criança; Relatos de casos
CASE REPORTS RELATOS DE CASOS
Embryonal rhabdomyosarcoma of the caruncle in a 4 year-old boy: case report
Rabdomiossarcoma embrionário da carúncula em um menino de 4 anos: relato de caso
Maria Del Carmen MendezI; Yerena MuiñosII; Gonzalo BlancoIII; Maria Antonia SaornilIV; Ciro García-AlvarezV; Jose Luis SarasaVI; Carmen ValbuenaVII
IPhysician, Hospital Severo Ochoa, Servicio de Patología, Madrid, Spain
IIPhysician, Hospital POVISA, Unidad de Oculoplástica y Oncología Ocular, Vigo, Spain
IIIPhysician, Hospital Rio Hortega, Unidad de Oculoplástica, Clínica QUO, Valladolid, Spain
IVPhysician, Hospital Clínico Universitario de Valladolid, Unidad de Tumores Intraoculares, y Clínica QUO, Valladolid, Spain
VPhysician, Hospital Clínico Universitario de Valladolid, Unidad de Tumores Intraoculares, Valladolid, Spain
VIPhysician, Fundación Jimenez Díaz, Universidad Autónoma de Madrid, Madrid, Spain
VIIPhysician, Hospital Clínico Universitario de Valladolid, Servicio de Pediatría, Valladolid, Spain
ABSTRACT
Rhabdomyosarcoma is a rare tumor, with an annual incidence of 4.3 cases per million children. Even thought, it is the most common soft tissue sarcoma in childhood, with a mean age of 6 to 8 years at diagnosis. A 4 year-old boy presented with a history of a fast growing (1-month) nodular lesion in the caruncle of his left eye. Slit lamp examination showed a vascularized solid nodular lesion in the semilunar fold. The lesion was surgically removed obtaining infiltrated edges with tumoral cells. A second surgery was performed with free tumour edges. The diagnosis of embryonal rhabdomyosarcoma, botryoid type, of intermediate differentiation was made. The treatment for botryoid rhabdomyosarcoma is basically surgical with the combined use of adjuvant polychemotherapy. In adolescent or adult patients (not in infants where growth bone disturbances can occur) external beam radiotherapy can be combined with chemotherapy. Rhabdomyosarcomas of the conjunctiva without orbital extension are rarely reported. We presented a case of a child with a rare tumor which we had a high suspicion of malignancy and early diagnosis and treatment and child is free of systemic disease 6 and half years later. Our research group believes that the key in these tumors is the high index of suspicion and early treatment.
Keywords: Rhabdomyosarcoma/surgery; Conjuctival neoplasms; Rhabdomyosarcoma, embryonal/surgery; Humans; Male; Children; Case report
RESUMO
Rabdomiossarcoma é um tumor raro, com uma incidência anual de 4,3 casos por milhão de crianças. É o sarcoma de partes moles mais comum na infância, com uma idade média de 6 a 8 anos no momento do diagnóstico. Um menino de 4 anos apresentou-se com uma história de um crescimento rápido (1 mês) lesão nodular em carúncula de seu olho esquerdo. O exame biomicroscópico mostrou uma lesão vascularizada sólida nodular na prega semilunar. A lesão foi removida cirurgicamente com obtenção de bordas infiltradas com células tumorais, segunda cirurgia foi feita com bordas tumorais livres. O diagnóstico de rabdomiossarcoma embrionário, tipo botrióide, de diferenciação intermediária foi feita. O tratamento para a rabdomiossarcoma botrióide é basicamente cirúrgico com o uso combinado de poliquimioterapia adjuvante. Em pacientes adolescentes e adultos (e não em lactentes onde os distúrbios do crescimento ósseo pode ocorrer) radioterapia externa pode ser combinada com a quimioterapia. Rabdomiossarcomas da conjuntiva sem extensão orbital são raramente relatadas. Apresentamos um caso de uma criança com um tumor raro que tivemos uma alta suspeita de malignidade e diagnóstico precoce e tratamento e criança é livre de doença sistêmica 6 anos e meio depois. Nosso grupo de pesquisa acredita que a chave para esses tumores é o alto índice de suspeita e tratamento precoce.
Descritores: Rabdomiossarcoma/cirurgia; Neoplasias da tunica conjuntiva; Rabdomiossarcoma embrionário/cirurgia; Humanos; Masculino; Criança; Relatos de casos
INTRODUCTION
Rhabdomyosarcoma is a rare tumor, with an annual incidence of 4.3 cases per million children(1). Even thought, it is the most common soft tissue sarcoma in childhood, with a mean age of 6 to 8 years at diagnosis(1). It can occur in any anatomic location of the body where there is skeletal muscle, as well as other sites without skeletal muscle, such as the soft tissues of the orbit. It is presumed that most of these lesions, particularly the embryonal variants, arise from undifferentiated or pluripotential mesenchyme. The primary sites of rhabdomyosarcoma include(2) head and neck (40%), genitourinary tract (20%), extremities (20%), trunk (10%) and others (10%). In the eye, it can affect orbit, eyelids, conjunctiva and uveal tract(3-5). Rhabdomyosarcomas of the conjunctiva without orbital extension are rarely reported(2,6). We presented a case of a child with a rare tumor which we had a high suspicion of malignancy and early diagnosis and treatment, and child is free of systemic disease 6 and half years later.
CASE REPORT
A 4 year-old boy presented in our institution with a history of a fast growing (1-month) nodular lesion in the caruncle of his left eye. Slit lamp examination showed a vascularized solid nodular lesion in the semilunar fold (Figure 1). Otherwise ocular examination was unremarkable and no other conjunctival or ocular lesions were observed. Systemic studies were negative.
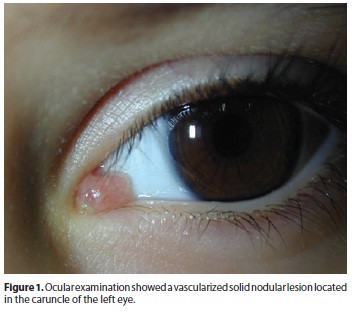
Familial history showed a cousin died due to a rhabdomyosarcoma of the biliary tract.
The lesion was surgically removed obtaining infiltrated edges with tumoral cells, second surgery was made with free tumor edges. Adjuvant polichemotherapy treatment, following the Protocol of the International Society of Pedriatric Oncology for Malignant Mesenchymal Tumors, was performed using ifosfamide, vincristine, and actinomycin (IVA) - vincristine (V) in alterned cicles for 7 cicles.
RESULTS
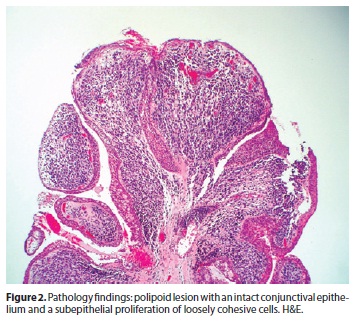
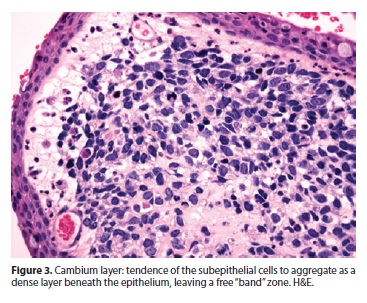
A polipoid lesion, with an intact conjunctival epithelium was seen (Figure 2). There were tumoral cells in the substantia propia, and in some areas they were aggregated as a denser layer beneath the epithelium (Figure 3). At higher magnification the undifferentiated tumor was composed of loosely coherent spindle-shaped cells, some of them with red-staining citoplasms, oriented in various directions. The cellular atypia and pleomorphism were evident, and there was a high proliferative rate (60-70%). Cross striations were not readily visualized. There was an image highly suggestive of lymphatic invasion.
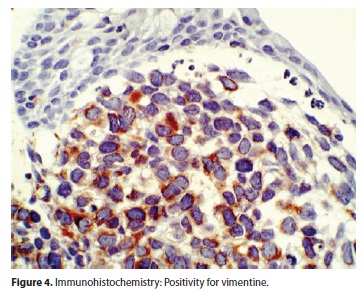
On immunohistochemistry many tumour cells showed strong reactivity with vimentine (Figure 4), muscle-specific actin and desmin antibodies and to a lesser extent with an antibody against Myo-D1.
The diagnosis of embryonal rhabdomyosarcoma, botryoid type, of intermediate differentiation was made.
DISCUSSION
Rhabdomyosarcoma originating in the conjunctiva show a propensity to arise in the superior or superonasal areas of the conjunctival cul-de-sac, but they also may arise in other quadrants. The initial clinical manifestation of the tumor may be as a noninflamed pedicle of soft tissue originating in the fornix and protruding into the palpebral aperture. A botryoid rhabdomyosarcoma clinically does not necessarily show rapid growth.
Based on histopathologic observation, these tumors have been subdivided into embryonal (83%), alveolar (15%), and pleomorphic ("adult type") forms (2%). Patient survival correlate with the morphologic subtype at diagnosis(7)(embryonal rhabdomyosarcoma has the more favorable prognosis1). CT and MR imaging are important in the evaluation of this tumor but final histophthological diagnosis is based in immunohistologic reactivity for desmin and myoglobin, two muscle-related proteins(6).
Rhabdomyosarcoma may also be encountered in the rare Li-Fraumeni syndrome, in which there is a predisposition to the development of various neoplasia, including sarcoma, breast carcinoma, central nervous system tumors, germ cell tumors, and leukemia. A mutation of the p53 suppressor gene has been identified as the causative factor. Patients with Li-Fraumeni syndrome not only present with neoplasia at an earlier age and have a worse prognosis than does the general population, but also have an increased risk for the development of secondary malignancies within the radiation field.
The treatment for botryoid rhabdomyosarcoma is basically surgical with the combined use of adjuvant polychemotherapy. In adolescent or adult patients (not in infants where growth bone disturbances can occur) external beam radiotherapy can be combined with chemotherapy. Some authors(1), due to the post-radiation complications, propose to use radiotherapy only in cases with bad response to chemotherapy(1). There is an innovative therapeutic approach for a rhabdomyosarcoma with brachytherapy with strontium-904, whose advantage is the high surface dose combined with a minimal depth of irradiation.
Our research group believes that the key in these tumors is the high index of suspicion and the early treatment, and in our knowledge there is no publish report of such extremely rare case with favorable evolution.
REFERENCES
1. Karcioglu ZA, Hadjistilianou D, Rozans M, DeFrancesco S. Orbital rhabdomyosarcoma. Cancer Control. 2004;11(5):328-33.
2. Brichard B, De Potter P, Godfraind C, Vermylen C. Embryonal rhabdomyosarcoma presenting as conjunctival tumor. J Pediatr Hematol Oncol. 2003;25(8):651-2.
3. Shields CL, Shields JA, Honavar SG, Demirci H. Clinical spectrum of primary ophthalmic rhabdomyosarcoma. Ophthalmology. 2001;108(12):2284-92.
4. Sekundo W, Roggenkamper P, Fischer HP, Fleischhack G, Fluhs D, Sauerwein W. Primary conjunctival rhabdomyosarcoma: 2.5 years' follow-up after combined chemotherapy and brachitherapy. Graefes Arch Clin Exp Ophthalmol. 1998;236(11):873-5.
5. Shields CL, Shields JA, Honavar SG, Demirci H. Primary ophthalmic rhabomyosarcoma in 33 patients. Trans Am Ophthalmol Soc. 2001;99:133-42; discussion 142-3.
6. Cameron JD, Wick MR. Embryonal rhabdomyosarcoma of the conjuctiva. A clinicopathologic an immunohistochemical study. Arch Ophthalmol. 1986;104(8):1203-4.
7. Kodet R, Newton WA Jr, Hamoudi AB, Asmar L, Wharam MD, Maurer HM. Orbital rhabdomyosarcomas and related tumors in childhood: relationship of morphology to prognosis-an Intergroup Rhabdomyosarcoma study. Med Pediatr Oncol. 1997; 29(1):51-60.
 Correspondence address:
Correspondence address:
Maria Del Carmen Mendez Diaz, MD.
Hospital Severo Ochoa
Servicio de Patología - Avda de Orellana s/n - Leganés Madrid
E-mails: [email protected]/[email protected]
Submitted for publication: September 27, 2011
Accepted for publication: January 19, 2012
Study carried out at Hospital Clínico Universitario de Valladolid colaborated with Hospital Povisa - Hospital Rio Hortega.
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: M.D.C.Mendez, None; Y.Muiños, None; G.Blanco, None; M.A.Saornil, None; C.García-Alvarez, None; J.L.Sarasa, None; C.Valbuena, None.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.