Arq. Bras. Oftalmol. 2001; 64 (3): 10.1590/S0004-27492001000300017
Total: 2118
Agostinho Bryk Junior; Maria Claudia Gomes Komatsu; Silvane Bigolin; Leciana Rorato Chiconelli Vanzo1; Eduardo Luis Jansson1; Ana Tereza Ramos Moreira2
DOI: 10.1590/S0004-27492001000300017
RESUMO
A osteopetrose congênita é uma rara desordem genética autossômica recessiva caracterizada por osso esclerótico associado a anormalidades hematológicas e neurológicas. Os autores fazem revisão da literatura e relatam um caso de uma criança do sexo feminino com 2 anos e 5 meses de vida apresentando amaurose bilateral por osteopetrose congênita.
Descritores: Osteopetrose; Osteopetrose; Cegueira; Relato de caso; Criança
ABSTRACT
Osteopetrosis is a rare autosomal recessive genetic disorder characterized by sclerotic bones associated with hematologic and neurologic abnormalities. The authors review the literature and report a case of a girl aged 2 years and 5 months who presented bilateral amaurosis due to congenital osteopetrosis.
Keywords: Osteopetrosis; Osteopetrosis; Blindness; Case report; Child
INTRODUÇÃO
Em 1904, Heinrich Albers Schönberg, radiologista alemão, relatou o primeiro caso de osteopetrose em um homem de 26 anos com esclerose óssea generalizada e múltiplas fraturas(1-5). Nesta mesma época também foi denominada doença marmórea ou osteosclerosis fragilis generalisata(1, 4).
Trata-se de doença rara, de caráter hereditário e que pode se manifestar de várias formas e em qualquer idade. Sua etiopatogenia ainda não está bem definida, não havendo predomínio por raça, porém acomete ligeiramente mais o sexo masculino(2-3). Segundo Barba e col.(6), aproximadamente 20% dos casos derivam de casamentos consangüíneos.
A fisiopatologia é explicada por um defeito básico na diferenciação das células precursoras em osteoclastos. O osso contém número aumentado de osteoclastos estruturalmente anômalos(1).
São comuns as infecções pois os neutrófilos apresentam resposta quimiotática diminuída e baixa capacidade de fagocitose bacteriana(4).
A osteopetrose classifica-se clinicamente em dois grupos: congênita (maligna) - transmitida por padrão autossômico recessivo - e tardia (benigna) - autossômica dominante(1-3). Alguns autores consideram ainda a forma intermediária(4).
O presente caso refere-se à forma autossômica recessiva ou congênita. A osteopetrose congênita ocorre numa incidência de 1/500.000 habitantes nos Estados Unidos e de 1/200.000 no Brasil(4). Nesta forma as manifestações clínicas são evidentes durante a infância, podendo se fazer o diagnóstico intra-uterino ou nos primeiros anos de vida(7). Há supercrescimento ósseo com disfunção medular, fraturas espontâneas, anemia, plaquetopenia, hepatoesplenomegalia, enfartamento ganglionar e aumento do timo(1, 3, 5).
Em estágios mais avançados da doença podem ocorrer paralisias dos nervos óptico, oculomotor e facial pela compressão resultante do supercrescimento ósseo(1, 4). O nervo óptico é o mais comumente afetado e o facial fica em segundo lugar, cuja disfunção pode ser o primeiro sinal da osteopetrose(8).
O comprometimento neurológico ainda pode se manifestar com hidrocefalia, trombose do seio sagital, exoftalmia, oclusão do forame da base do crânio comprimindo nervos e vasos, resultando em paresia, surdez e amaurose(1).
O óbito geralmente ocorre por infecção ou sangramento na 1ª década de vida(1, 8).
O estudo radiológico demonstra esclerose óssea generalizada. Os focos de esclerose que ocorrem nas vértebras, pélvis e ossos longos distais são os aspectos patognomônicos(1, 2, 6, 8). Nota-se ainda espessamento da base e da calota craniana, perda da díploe e diminuição ou ausência do canal medular(8).
Os exames laboratoriais demonstram pancitopenia com hematócrito baixo em casos graves(2). Há um aumento da fosfatase ácida e as taxas de cálcio, fósforo e fosfatase alcalina são normais na grande maioria dos pacientes(2).
O tratamento mais aceito e em uso na última década é o transplante de medula óssea(1-9).
MÉTODOS
M.R.M., nascida em 21/01/1998, sexo feminino, branca, natural e procedente de Marechal Cândido Rondon (PR).
Paciente encaminhada ao serviço apresentando amaurose bilateral. Mãe relatou que a criança apresentava nistagmo e não seguia objetos desde os 3 meses de idade.
Em seu desenvolvimento psicomotor, apresentou sustentação cefálica e sentou sem apoio aos 5 meses e anda com andador. Iniciou fala com significado aos 10 meses.
Pais saudáveis e sem história de consangüinidade. Filha única, parto natural, a termo, peso de 3650g. Gestação e parto sem intercorrências.
Ao exame físico apresenta peso e estatura baixos para a idade. Bom estado geral, hipocorada e atitude ativa. Há macrocrania, protusão frontal e má formação dentária.
Ao exame oftalmológico apresenta nistagmo rotatório, motilidade intrínseca ausente. Paciente não fixa nem segue objetos. A pressão intra-ocular, a biomicroscopia e a gonioscopia são normais. O fundo de olho (Figura 1) apresenta papila atrófica bilateral e ausência de fixação macular.
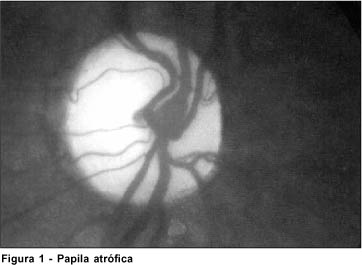
Na avaliação neurológica observa-se desenvolvimento cognitivo adequado para a idade e discreto atraso motor. Há perda auditiva moderada bilateral.
A radiografia de crânio (Figura 2) e membros (Figura 3) demonstra importante esclerose difusa e uniforme, arquitetura totalmente obliterada da calota craniana, ossos da base do crânio, coluna cervical e dos membros inferiores. Tomografia axial computadorizada e ressonância nuclear magnética normais. Leucocitose, trombocitopenia e anemia na avaliação da hematologia pediátrica.
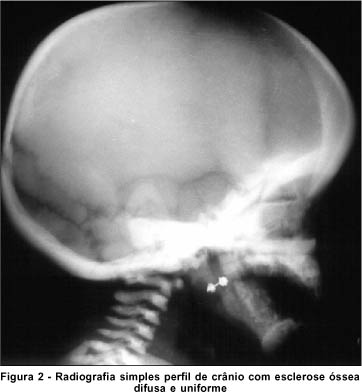
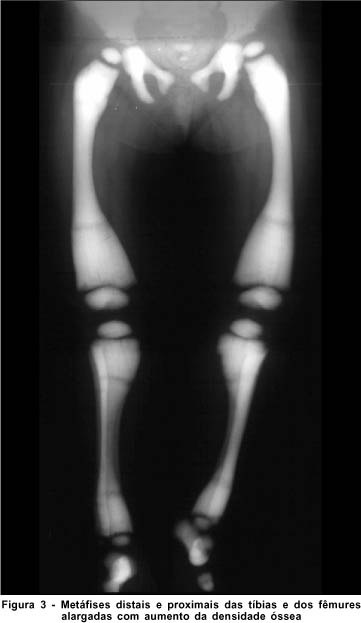
DISCUSSÃO
O caso relatado apresenta achados radiológicos característicos da doença, além da apresentação clínica e laboratorial muito próximas às da literatura.
Segundo Ainsworth e col.(7), seria apropriado investigar especificamente a osteopetrose em todas as crianças que apresentem redução visual nos primeiros meses de vida, na ausência de uma causa óbvia. Os achados oculares característicos são atrofia óptica, nistagmo, paralisia do nervo abducente e a obstrução do ducto nasolacrimal. A palidez do disco óptico está sempre presente, embora em grau variado.
Algumas teorias têm sido propostas para explicar a perda visual na osteopetrose, sendo elas a compressão óssea, a desmielinização do nervo óptico, o edema de papila e a degeneração retiniana(7). Embora a compressão do nervo óptico tenha sido considerada a principal causa de atrofia óptica e conseqüente amaurose na osteopetrose, alguns autores têm sugerido a degeneração retiniana como o principal fator em alguns casos(9). Esta evidência pode ser comprovada pela melhora da função visual e neurológica observada em certos pacientes após o transplante de medula óssea(9).
De acordo com Jinkins(10), pacientes que apresentam diminuição da acuidade visual mas sem atrofia óptica severa devem ser submetidos à descompressão cirúrgica do nervo óptico a fim de se evitar a progressão da lesão nervosa. No entanto Angeli e col.(8) propõem que há uma propensão para atrofia do nervo óptico mesmo com mínima manipulação durante a descompressão cirúrgica, tendo sido frustrante, portanto, este procedimento.
O transplante de medula óssea HLA idêntico representa a melhor opção de tratamento, pois pode promover a remissão completa da doença(7). Contudo deve ser realizado o mais precoce possível pelo risco de infecção recorrente e também pelo fato de que, com a idade, aumentam os riscos de complicações pós-transplante como, por exemplo, a hipercalcemia(7). Além disso não existe medicamento capaz de conter a doença(8). Não havendo tratamento adequado há um alto índice de morbidade e mortalidade, sendo que poucas crianças sobrevivem até a adolescência(1).
A paciente atualmente aguarda o transplante, necessitando de uma medula óssea HLA compatível do Banco Internacional de medula óssea.
CONCLUSÃO
É importante realizar o diagnóstico da osteopetrose congênita o mais precoce possível a fim de planejar a conduta terapêutica mais apropriada e tentar evitar as complicações decorrentes da evolução da doença. Infelizmente as opções de tratamento não vêm se mostrando eficazes com exceção do transplante de medula óssea que tem mostrado bons resultados em alguns casos.
REFERÊNCIAS
1. Dias ML, Rocha ML, Wai PA, Lizo CL. Osteopetrose congênita - relato de um caso. Rev Paul Pediatr 1984;2:33-6.
2. Jeronymo JRB, Borba LAN. Osteopetrose - doença de Albers-Schönberg. Relato de um caso. Radiol Bras 1989;22:137-40.
3. Salomão Jr JB, Menin AMCR, Baldi HS, Simone VMP de. Osteopetrose: relato de um caso. Rev Paul Pediatr 1997;15:47-51.
4. Colônia AMN, Schaimberg CG, Yoshinari NH, Santos M, Jorgetti V, Cossermelli W. Osteopetrose: relato de dois casos e revisão da literatura. Rev Hosp Clin Fac Med Univ S Paulo 1993;48:242-7.
5. Vilanova LCP, Pereira AMD, Carvalho D de S, Morgulis RF. Osteopetrose ou doença de Albers-Schönberg - Relato de um caso. Rev Paul Pediatr 1988; 6:106-8.
6. Barba MF, Doria AS, Torre MB, Nakano EK, Kim CA, Andrade MR, Bogus LCN, Scatigno Neto A. Osteopetrose - Relato de dois casos e diagnóstico por imagem. Pediatria (São Paulo) 1995;17:60-3.
7. Ainsworth JR, Bryce IG, Dudgeon J. Visual loss in infantile osteopetrosis. J Pediatr Ophthalmol Strabismus 1993;30:201-3.
8. Angeli CB, Flamia CL, Amoretti RK, Prado DA do, Linden A. Osteopetrose: relato de caso. Rev Bras Otorrinolaringol 1998;64:73-5.
9. Walpole IR, Nicoll A, Goldblatt J. Autosomal dominant osteopetrosis type II with "malignant" presentation: further support for heterogeneity? Clin Genet 1990;38:257-63.
10. Jinkins JR. The optic neurogram: evaluation of CSF "block" caused by compressive lesions at the optic canal. AJNR Am J Neuroradiol 1987;8:135-9.
11. Longhitano SB, Brunoni D, Lederman HM, Coelho MJRG. Osteopetrosis: a case report. Rev Hosp São Paulo Esc Paul Med 1990;2:89-90.
12. Siatkowski RM, Vilar NF, Sternau L, Coin CG. Blindness from bad bones. Surv Ophthalmol 1999;43:487-90.
Trabalho realizado no Serviço de Oftalmologia do Hospital de Clínicas da Universidade Federal do Paraná.
1 Acadêmicos do 5º ano de medicina.
2 Acadêmica do 6º ano de medicina.
3 Médicos Residentes em Oftalmologia do Hospital de Clínicas da Universidade Federal do Paraná.
4 Chefe do serviço de Oftalmologia da Universidade Federal do Paraná.
Os autores declaram não possuir interesse financeiro no desenvolvimento ou marketing de qualquer instrumento ou substância referidos no estudo.
Endereço para correspondência: Maria Claudia Gomes Komatsu. Rua Alferes Poli, 271 - Curitiba (PR) CEP 80230-090. E-mail: [email protected]
Nota Editorial: Pela análise deste trabalho e por sua anuência sobre a divulgação desta nota, agradecemos a Dra. Rosana Pires N. da Cunha.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.