Arq. Bras. Oftalmol. 2011; 74 (4): 10.1590/S0004-27492011000400014
Total: 2763
Marcelo Mendes Lavezzo1; Alan Kardec Barreira Jr1; Leandro Cabral Zacharias2; Walter Yukihiko Takahashi2
DOI: 10.1590/S0004-27492011000400014
RESUMO
O objetivo é relatar o caso de um paciente de sete anos, nascido a termo, sem intercorrências perinatais, encaminhado ao Setor de Retina/Vítreo para elucidação diagnóstica. Apresentava história de redução da acuidade visual à esquerda, de caráter insidioso/progressivo, há quatro anos. Ao exame, apresentava diminuição do diâmetro corneano e corectopia do olho direito (OD), sem alterações à biomicroscopia do olho esquerdo (OE). A fundoscopia do OD revelava descolamento de retina (DR) total e, do OE, inicialmente, mostrava alterações vasculares retinianas periféricas e exsudação retiniana, associado à tração vitreorretiniana no setor temporal. As tomografias e ressonâncias de crânio/órbitas não apresentavam anormalidades, com exceção de achados sugestivos de DR antigo no OD, confirmado pela ultrassonografia do globo ocular, que também demonstrou microftalmia. Diante disso, aventou-se a hipótese diagnóstica de vitreorretinopatia exsudativa familiar, doença rara de caráter autossômico dominante e relacionada com casamentos consanguíneos, inicialmente simulando doença de Coats. O paciente foi tratado com fotocoagulação a laser diodo na periferia temporal do OE, com melhora das áreas de tração vitreorretiniana.
Descritores: Vitreorretinopatia proliferativa; Oftalmopatias hereditárias; Descolamento retiniano; Criança; Relato de caso
ABSTRACT
We report the case of a seven year-old male patient, born at term without any perinatal complications, referred to the Retina/Vitreous Service for diagnostic elucidation. He had a history of progressive visual acuity loss on his left eye that started four years ago. On examination, he had decreased corneal diameter and corectopia of the right eye (OD), without any noteworthy findings on the biomicroscopy of the left eye (OS). The fundus of the OD revealed total retinal detachment, and the OS initially showed peripheral retinal vascular abnormalities and retinal exudation, associated with retinal vitreous traction on the temporal sector. The CT and MRI of the brain/orbits showed no abnormalities, except for findings suggestive of an old retinal detachment on the OD, confirmed by ultrasonography, which also showed microphthalmia of the OD. The diagnosis of familial exudative vitreoretinopathy, a rare disease of autosomal dominant inheritance and related to consanguineous marriages, that can initially simulate Coats disease, was proposed. The patient was treated with diode laser photocoagulation in the temporal periphery of the OS, with improvement in the areas of vitreoretinal traction.
Keywords: Vitreoretinopathy, proliferative; Eye diseases, hereditary; Retinal detachment; Child; Case report
INTRODUÇÃO
A vitreorretinopatia exsudativa familiar (FEVR) é uma doença caracterizada pelo desenvolvimento anormal da retina em crianças nascidas a termo, fazendo parte do amplo espectro das retinopatias exsudativas e das desordens vasculares retinianas.
Os principais diagnósticos diferenciais são: doença de Coats, retinopatia da prematuridade, fibroplasia retrolental, dobras retinianas, entre outros(1).
O objetivo é relatar um caso de FEVR em um menino de sete anos de idade simulando doença de Coats, além de descrever a evolução diagnóstica para esta condição rara.
RELATO DE CASO
Paciente de sete anos, masculino, pardo, encaminhado ao Setor de Retina/Vítreo do HC-FMUSP para elucidação diagnóstica. A mãe da criança referia que, desde o nascimento, a mesma apresentava nistagmo, com aparecimento de exotropia com desvio do olho direito (OD) aos 2 anos. Relatava redução da acuidade visual (AV) do olho esquerdo (OE), de caráter progressivo/insidioso, desde os três anos de idade. Nascido a termo, 2.700 g, sem intercorrências perinatais, não necessitando de oxigenioterapia. Seus pais são consanguíneos (primos de primeiro grau).
Ao exame, apresentava AV de sem percepção luminosa (SPL) no OD e de 20/125 (refração: -1,75DE // -2,00DCx170º) no OE; exotropia preferindo OE; microftalmia do OD, além de nistagmo com aumento de frequência/amplitude às dextro/levoversões. À biomicroscopia do OD apresentava: ceratopatia em faixa, câmara anterior sem reação, corectopia, sem rubeosis iridis. Apresentava pressão intraocular de 4 mmHg (OD) e de 16 mmHg no OE. A fundoscopia do OD revelava descolamento de retina (DR) total seroso e o OE mostrava disco óptico elevado com bordas pouco nítidas, retina aplicada, vasos com aspecto de tração para região temporal, alteração vascular na periferia temporal com traves vítreas neste setor (Figura 1).
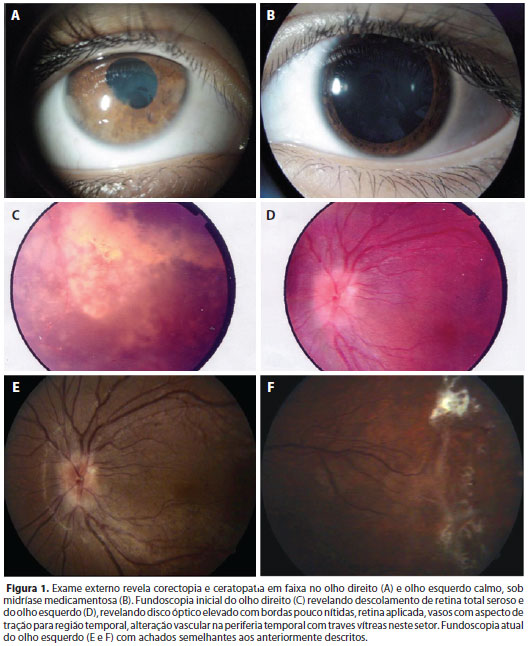
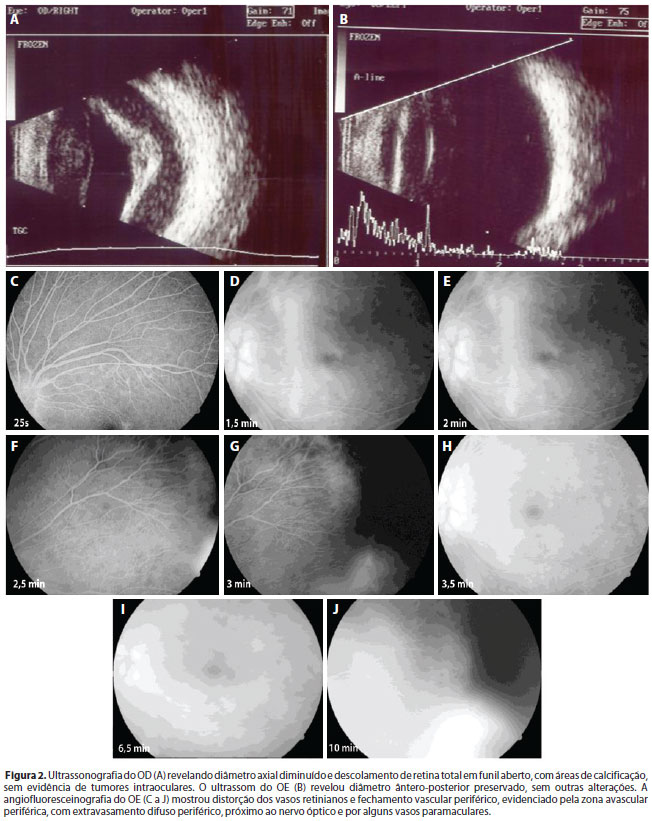
A ultrassonografia do OD revelou diâmetro axial diminuído e DR total em funil aberto, com áreas sugestivas de calcificação, sem evidência de tumores intraoculares. A angiofluoresceinografia do OE revelava distorção dos vasos retinianos e oclusão vascular periférica, com extravasamento difuso periférico, próximo ao nervo óptico e por alguns vasos paramaculares (Figura 2).
As tomografias computadorizadas e ressonância nuclear magnética de crânio/órbitas, sorologias (rubéola, sífilis, toxoplasmose e hepatites B/C), prova de falcização e análise do líquor estavam normais.
O exame oftalmológico dos pais/irmãos do paciente era normal (exceção: esotropia no irmão mais velho).
Com relação ao OD, optou-se por observação, devido ao tempo de história/aspecto isquêmico da retina, além da AV de SPL e da hipotonia. Quanto ao OE, optou-se por aplicação de fotocoagulação com laser diodo com objetivo de ablação da retina isquêmica da periferia temporal, havendo melhora das traves vítreas neste setor. O paciente mantém seguimento ambulatorial até o presente momento, apresentando AV estável e melhora da exsudação lipídica e da tração temporal.
DISCUSSÃO
A FEVR é uma doença autossômica dominante (cromossomo: 11q), de traço recessivo ligado ao X ou esporádica(2). Apresenta acometimento bilateral e assimétrico, variando de uma forma assintomática e não-progressiva até uma forma proliferativa ativa (mais comum). Acomete ambos os sexos, sendo diagnosticada, usualmente, na infância(1).
Representa uma causa significativa de DR em pacientes com menos de 30 anos(3,4), sendo um dos diagnósticos diferenciais de DR na infância.
As alterações vasculares retinianas são semelhantes às da retinopatia da prematuridade, todavia os indivíduos acometidos apresentam história de nascimento a termo, com peso ao nascimento adequado, sem história de intercorrências perinatais (inclusive sem necessidade de oxigenioterapia)(1).
A fundoscopia geralmente apresenta: tração retiniana temporal (dragging), zona avascular na retina periférica e vasos retinianos periféricos retificados formando anastomoses ao longo da interface avascular. Pode haver evolução destas lesões com proliferação e exsudatos/hemorragias sub-retinianos, além de DR e glaucoma neovascular(1).
A angiofluoresceinografia caracteriza-se por: extravasamento do contraste dos vasos anormais com terminações aneurismáticas na interface vascular-avascular da retina periférica. A zona avascular pode estar presente nos 360º periféricos, entretanto a zona de isquemia caracteristicamente é mais ampla no setor temporal(1).
Gow e Oliver(5) propuseram uma classificação da FEVR em três estadiamentos clínicos. Há também outras classificações propostas, de acordo com a gravidade da doença, tipo/extensão do DR e seu componente (predominantemente exsudativo/tracional)(4,6). Apesar disso, sabe-se que há uma grande variabilidade na sua expressão fenotípica, visto que uma parte considerável de pacientes apresenta uma forma moderada da doença, que só pode ser detectada com o auxílio da angiofluoresceinografia. Em uma série de 12 casos de FEVR, embora alguns fossem assintomáticos (7 pacientes: 14 olhos), a angiofluoresceinografia demonstrou que praticamente todos apresentavam alteração na perfusão da retina periférica (13 olhos), sem outros achados, ratificando mais uma vez sua importância no arsenal diagnóstico(3).
Há poucos casos de FEVR descritos na literatura nacional(7,8), embora esta doença possa ser mais comum do que o relatado, devido à sua dificuldade diagnóstica e considerável número de diagnósticos diferenciais.
Diante dos exames, as principais hipóteses diagnósticas aventadas foram: FEVR, doença de Coats e retinopatia da prematuridade. Entretanto, esta é menos provável, pela ausência de intercorrências perinatais.
Por sua vez, a doença de Coats é caracterizada por telangectasias retinianas idiopáticas com exsudação intra/sub-retiniana, sem tração retiniana/vítrea aparentes. É uma condição esporádica, nãohereditária, que não está associada a anormalidades sistêmicas. Em uma revisão de 150 casos, verificou-se que a média de idade do diagnóstico foi de 11 anos, em indivíduos brancos (82%), do sexo masculino (76%), de acometimento unilateral (95%) e predominantemente do quadrante temporal (42%)(9).
Embora haja algumas semelhanças, na FEVR, não há preferência por sexo e o acometimento é bilateral. Assim, a constatação de tração vitreorretiniana no setor temporal e os achados sugestivos da angiofluoresceinografia corroboraram sobremaneira para o diagnóstico de FEVR.
Pelo fato da apresentação clínica desta doença ser tão assimétrica, esta acabou simulando doença de Coats inicialmente. Além disso, estudos recentes teorizam que mutações no gene NDP estariam associadas com um espectro de alterações retinianas, englobando desde a FEVR até a doença de Coats. Desse modo, cogita-se que estas doenças seriam expressões fenotípicas diferentes de alterações no mesmo gene. O NDP seria responsável pela codificação da proteína norrina, relacionada à angiogênese/desenvolvimento vascular da retina e estímulo direto na proliferação das células ganglionares. Os testes genéticos moleculares do gene NDP, embora clinicamente disponíveis(10), ainda não são factíveis em nosso meio. Assim, métodos diagnósticos auxiliares adicionais são essenciais, bem como um exame pormenorizado, para melhor caracterização/diferenciação entre estas duas patologias.
Os tratamentos propostos para a FEVR consistem em observação, fotocoagulação periférica a laser (estágios iniciais) e/ou cirurgia vitreorretiniana (casos avançados bem selecionados)(5). Como os resultados anatômicos/funcionais são menos promissores em olhos com estágios mais avançados de FEVR, uma intervenção precoce parece ser benéfica.
Glaucoma neovascular e DR são complicações associadas à FEVR(1). Além disso, existe a possibilidade de rápida progressão da doença no olho menos acometido. Por conseguinte, é essencial o conhecimento desta doença e de seus diferenciais para que seja feito um diagnóstico precoce e o tratamento seja prontamente instituído antes do desenvolvimento do DR extenso. Desse modo, recomenda-se que os pacientes afetados sejam examinados regularmente, bem como seja realizado o exame preventivo de seus familiares para se detectar casos de FEVR em estágios precoces.
REFERÊNCIAS
1. Tasman W, Augsburger JJ, Shields JA, Caputo A, Annesley WH Jr. Familial exudative vitreoretinopathy. Trans Am Ophthalmol Soc. 1981;79:211-26.
2. Plager DA, Orgel IK, Ellis FD, Hartzer M, Trese MT, Shastry BS. X-linked recessive familial exudative vitreoretinopathy. Am J Ophthalmol. 1992;114(2):145-8.
3. Ober RR, Bird AC, Hamilton AM, Sehmi K. Autosomal dominant exudative vitreoretinopathy. Br J Ophthalmol. 1980;64(2):112-20.
4. Miyakubo H, Hashimoto K, Miyakubo S. Retinal vascular pattern in familial exudative vitreoretinopathy. Ophthalmology. 1984;91(12):1524-30.
5. Gow J, Oliver GL. Familial exudative vitreoretinopathy. An expanded view. Arch Ophthalmol.1971;86(2):150-5.
6. Pendergast SD, Trese MT. Familial exudative vitreoretinopathy. Results of surgical management. Ophthalmology. 1998;105(6):1015-23.
7. Portella E, Ramos ARB, Moreira JCA, Moreira ATR. Manejo da vitreoretinopatia exsudativa familial. Rev Bras Oftalmol. 1995;54(8):565-8.
8. Biccas Neto L, Mesquita AS, Louro ID. Familial exudative vitreoretinopathy (FEVR) associated with infantile osteoporosis: case report. Arq Bras Oftalmol. 2009;72(2):257-60.
9. Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001;131(5):561-71.
10. Sims KB. NDP-Related Retinopathies. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-1999 Jul 30 [updated 2009 Jul 23] .
Correspondence address:
Marcelo Mendes Lavezzo
Rua Capote Valente, 640 - Ap. 23
São Paulo (SP) - 05409-002 - Brazil
E-mail: [email protected]
Submitted for publication: December 21, 2009
Accepted for publication: May 15, 2010
Funding: No specific financial support was available for this study.
Disclosure of potential conflicts of interest: M.M.Lavezzo, None; A.K.Barreira Jr., None; L.C. Zacharias, None; W.Y.Takahashi, None.
Study carried out at the Clínica Oftalmológica do Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo - USP - São Paulo (SP), Brazil.
Editorial Note: After completing the confidential analysis of the manuscript, ABO discloses, with his agreement, the name Dr. João Borges Fortes Filho as a reviewer. We thank his effort and expertise in participating in this process.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
{kind=link}