Arq. Bras. Oftalmol. 2010; 73 (5): 10.1590/S0004-27492010000500016
Total: 1654
João Borges Fortes Filho1; Andrea Cunha Magnani2; Ana Paula Tonietto2; Fernanda Verçoza Lovato2; Humberto Lubisco Filho3
DOI: 10.1590/S0004-27492010000500016
ABSTRACT
Clinicopathologic findings of a 44-year-old Caucasian male who presented with a congenital bilateral chorioretinal coloboma and later developed a large choroidal melanoma in his best seeing eye. The eye containing the malignant tumor was enucleated and the histopathologic findings confirmed the clinical diagnosis of choroidal melanoma, and in this case, an epithelioid cell type.
Keywords: Coloboma; Eye abnormalities; Melanoma; Choroide neoplasms; Case reports
RESUMO
Achados clinicopatológicos de um paciente de 44 anos, branco, do sexo masculino, que apresentava coloboma bilateral congênito de retina e coróide e que, posteriormente, desenvolveu um grande melanoma da coróide no seu olho de melhor visão. O olho contendo o tumor maligno foi enucleado e os achados histopatológicos confirmaram o diagnóstico clínico de melanoma de coróide, neste caso do tipo celular epitelióide.
Descritores: Coloboma; Anormalidades do olho; Melanoma; Neoplasias da coróide; Relatos de casos
CASE REPORTS RELATOS DE CASOS
Choroidal melanoma in a patient with congenital bilateral chorioretinal coloboma: a case report of an extremely rare association of ocular comorbities
Melanoma de coróide em um paciente com coloboma congênito coriorretiniano bilateral: relato de caso de uma extremamente rara associação de comorbidades oculares
João Borges Fortes FilhoI; Andrea Cunha MagnaniII; Ana Paula ToniettoII; Fernanda Verçoza LovatoII; Humberto Lubisco FilhoIII
IPhysician, Medical School, Universidade Federal do Rio Grande do Sul - UFRGS - Porto Alegre (RS), Brazil
IIPhysician, Porto Alegre Eye Bank Hospital - Porto Alegre (RS), Brazil
IIIPhysician, Porto Alegre Eye Bank Hospital - Porto Alegre (RS), Brazil
ABSTRACT
Clinicopathologic findings of a 44-year-old Caucasian male who presented with a congenital bilateral chorioretinal coloboma and later developed a large choroidal melanoma in his best seeing eye. The eye containing the malignant tumor was enucleated and the histopathologic findings confirmed the clinical diagnosis of choroidal melanoma, and in this case, an epithelioid cell type.
Keywords: Coloboma/diagnosis; Eye abnormalities; Melanoma/secondary; Choroide neoplasms; Case reports.
RESUMO
Achados clinicopatológicos de um paciente de 44 anos, branco, do sexo masculino, que apresentava coloboma bilateral congênito de retina e coróide e que, posteriormente, desenvolveu um grande melanoma da coróide no seu olho de melhor visão. O olho contendo o tumor maligno foi enucleado e os achados histopatológicos confirmaram o diagnóstico clínico de melanoma de coróide, neste caso do tipo celular epitelióide.
Descritores: Coloboma/diagnóstico; Anormalidades do olho; Melanoma/secundário; Neoplasias da coróide; Relatos de casos.
INTRODUCTION
Colobomata are congenital malformations that result from an anomaly of the embryonic fissure. This anomaly can occur in the iris, lens, ciliary body, retina, choroid, and optic nerve. Retinochoroidal colobomas are frequently associated to numerous ocular and systemic anomalies, especially with retinal detachment in the affected eyes(1-2).
Choroidal melanomas are the most frequent primary malign intraocular tumors occurring much more commonly in white adults. The prevalence of this lesion reaches 5 or 7 cases per one million inhabitants per year(3).
This report describes the clinicopathologic findings of a patient who presented a congenital bilateral retinochoroidal coloboma and latter developed a large choroidal melanoma in his best seeing eye. The eye containing the malignant tumor was enucleated and the histopathologic findings disclosed a melanocytic epithelioid cell-type choroidal melanoma.
CASE REPORT
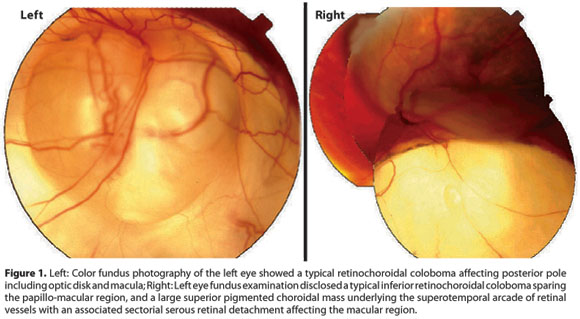
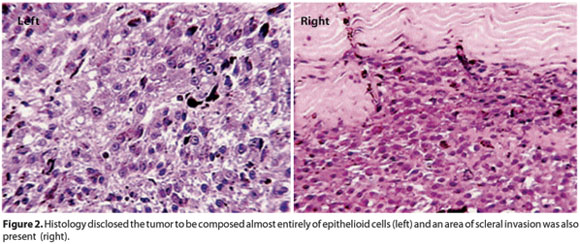
Approximately 3 years ago, a 44-year-old man presented with a complaint of blurred vision in his left eye (OS) for the past 30 days. The patient reported during his first visit that his left eye was always his best seeing eye. During the ophthalmic exam, both eyes presented low visual acuity, hand motion in OD, and counting fingers in OS. Anterior segment biomicroscopy was unremarkable in both eyes. Intraocular pressure measured by Perkins applanation tonometer was 18 mmHg in OD and 22 mmHg in OS. Fundus exam OD revealed a typical chorioretinal coloboma located inferiorly and extending from the periphery to the posterior pole also affecting the papillo-macular region. This finding explains the very low vision detected in this eye (Figure 1, right). Indirect ophthalmoscopy OS also disclosed a large chorioretinal coloboma inferiorly that spared the papillo-macular region. Furthermore, a large pigmented choroidal mass underlying the superotemporal vascular arcade of the retina with an associated serous retinal detachment was affecting the macular region of the OS. The clinical appearance of the mass was consistent with the diagnosis of choroidal melanoma. The lesion was located in the superior retina and far from the colobomatous area (Figure 1, left). Fluorescein angiography of the presumed tumor showed many described features of choroidal melanoma such as an independent vascular system or double-circulation; diffuse hyperfluorescence with blurred margins with leakage of dye from the vascular tumor network; many hyperfluorescent small spots (pin-points); late increase of leakage of dye and partial blocked fluorescence with irregular leakage into the subretinal space. The B-scan ultrasound of the OS revealed a large ocular mass consistent with choroidal melanoma measuring approximately 8.5 mm in thickness, 17 mm in basal vertical dimension, and 15.5 mm in basal horizontal dimension and with a suspicious image suggestive of scleral invasion by the tumor. Kappa-angle, suggestive for choroidal melanoma by echoes attenuation, was identified in the A-scan. The patient was referred to the Oncology Service where the hypothesis of metastatic dissemination of the circumscribed intraocular choroidal melanoma was ruled-out. After that, and due to the large size of the lesion coupled with the absence of any visual potential, enucleation was indicated. The eye containing the malignant tumor was enucleated with a good surgical outcome. After surgery, the macroscopic assessment of the enucleated globe revealed a 10 mm thick melanocytic mass associated with a serous retinal detachment. The basal size of the lesion was 17 mm by 16 mm. Tumor dimension findings were similar to the previous imaging study. Microscopic evaluation showed a tumor composed of approximately 95% epithelioid cells with scleral invasion as suggested by the ocular ultrasound (Figure 2).
A few weeks after enucleation, the patient was adapted with an external ocular prosthesis that presented a satisfactory cosmetic result, and was able to perform most of his daily tasks with the remaining subnormal vision of his right eye. Neither tumor recurrence nor metastatic disease was detected until July 2009.
DISCUSSION
Ocular coloboma is a congenital, common, and heterogeneous malformation which includes a spectrum of anomalies ranging from iris coloboma to clinical anophthalmos. As an isolated defect, it is usually inherited as an autosomal dominant disorder, although autosomal recessive inheritance also occurs. Patients with multiple malformations and coloboma may have a recognized malformation syndrome of unknown etiology, a single gene disorder, or chromosomal abnormality(2).
The typical coloboma occurs in the inferonasal quadrant, caused by defective closure of the fetal fissure. Congenital anomalies by defective embryogenesis can involve all layers of the eye and have numerous causes, most commonly of developmental genetic origin(4). Some patients with many malformations can have severely impaired vision, whereas others have only a cosmetic manifestation, and still others may have no symptoms and may go undiagnosed or may be detected incidentally on routine eye examination. Although chorioretinal colobomas may be totally asymptomatic, and may be present early in life, in most cases a hereditary pattern could not be established, as in the patient described herein.
Choroidal melanomas are known to be the most frequent primary malignant intraocular tumor occurring mainly in adults(3). Melanomas exhibit metastatic dissemination in the first five years after being diagnosed in approximately 50% of patients. The factors that are known risk factors for metastatic disease include the tumor size, ciliary body location, scleral and/or extra-scleral extension, histological cell type, among others. Large size choroidal melanomas not only have the potential to cause severe visual loss, but also high risk of metastasic disease to the liver, the lung, and rarely other organs(5-6). Once metastasis occurs, the death rate is 80% in the first year and 92% by two years(7).
Enucleation has been the treatment of choice for many years in eyes with large size choroidal melanomas. Eyes containing large lesions and no potential of visual recovery such as the one we described, do not have features that justify an attempt to salvage the eye. Because it is known that treating large tumors with radiation would require a high dose to the sclera and tumor base, eyes with an ocular malformation such as a coloboma would pose a higher risk of severe complications such as scleral necrosis and extra-scleral/orbital tumor extention(8).
We presented a rare association of bilateral congenital chorioretinal coloboma that affected the fovea and optic nerve in one eye, and that was associated with a large size choroidal melanoma occurring in the best seeing eye. To the best of our knowledge, this association has never been described before. We used literature search in Medline, PubMed and LILACS databases to search related clinical cases using keywords as choroidal and chorioretinal coloboma, and melanoma or choroidal melanoma, which leads us to assume that the association of both ocular comorbities in this patient most likely happened by chance. Furthermore, it seems very strange that in spite of both clinical entities as colobomas and melanomas being of frequent appearance in ophthalmic daily practice, their association in the same patient seems to be extremely rare.
REFERENCES
1. Pagon RA. Ocular coloboma. Surv Ophthalmol. 1981;25(4):223-36. Review.
2. Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Surv Ophthalmol. 2000;45(3):175-94.
3. Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology. 2003;110(5):956-61.
4. Guercio JR, Martyn LJ. Congenital malformations of the eye and orbit. Otolaryngol Clin North Am. 2007;40(1):113-40, vii. Review.
5. Shields JA, Shields CL, Donoso LA. Management of posterior uveal melanoma. Surv Ophthalmol. 1991;36(3):161-95.
6. Cheng KP, Hiles DA, Biglan AW. The differential diagnosis of leukokoria. Pediatr Ann. 1990;19(6):376-83, 386.
7. Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report nº 15. Arch Ophthalmol. 2001; 119(5):670-6. Comment in: Arch Ophthalmol. 2002;120(5):676.
8. Bedikian AY, Legha SS, Mavligit G, Carrasco CH, Khorana S, Plager C, et al. Treatment of uveal melanoma metastatic to the liver: a review of the M. D. Anderson Cancer Center experience and prognostic factors. Cancer. 1995;76(9):1665-70.
 Correspondence address:
Correspondence address:
João Borges Fortes Filho
Hospital Banco de Olhos de Porto Alegre
Rua Eng. Walter Boehl, 285
Porto Alegre (RS) - CEP 91360-090
E-mail: [email protected]
Recebido para publicação em 13.05.2008
Última versão recebida em 09.12.2009
Aprovação em 25.12.2009
Work carried out at the Retina Service - Ophthalmology Course, Porto Alegre Eye Bank Hospital - Porto Alegre (RS), Brazil.
Nota Editorial: Depois de concluída a análise do artigo sob sigilo editorial e com a anuência da Dra. Martha Maria Motono Chojniak sobre a divulgação de seu nome como revisora, agradecemos sua participação neste processo.

How to cite this article:
 ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.
ABO is licensed under a Creative Commons Attribution-NonComercial 4.0 Internacional.